
嘉峪检测网 2021-09-18 21:30
导读:生物仿制药与任何其他生物制品或原料药一样,都要经过同样严格的安全性和有效性评估。由于对原研药有较为多的知识积累和扎实的科学理解,因此生物仿制药与原研药的可比性证明会加快和简化生物仿制药的监管要求。仿制药的数据要求是根据具体情况与各国监管机构合作确定的。生物仿制药的可比性是基于风险的,比较的水平和复杂性与安全性和有效性的风险相称。此外,
生物仿制药与任何其他生物制品或原料药一样,都要经过同样严格的安全性和有效性评估。由于对原研药有较为多的知识积累和扎实的科学理解,因此生物仿制药与原研药的可比性证明会加快和简化生物仿制药的监管要求。仿制药的数据要求是根据具体情况与各国监管机构合作确定的。
可比性是多方面的,通常包括临床试验数据(药代动力学(pharmacokinetics,PK)概况)、动物体内药效学研究、有效成分子的分析可比性(功能和结构)、分析方法可比性(如果可能)、工艺可比性(批次间工艺对比研究),以及工艺过程控制和监测。在所有情况下的可比性都是原研药分子与评估的生物仿制药的比较。
生物仿制药的可比性是基于风险的,比较的水平和复杂性与安全性和有效性的风险相称。此外,使用基于风险的方法,证明可比性的统计方法同样可以分为三个层次进行比较。
可比性的三个层次
将可比性方法分为三个不同的层次或者类别是很有用的。第一层是最严格的基础比较,通常用于关键质量属性(CQA)、临床性能以及结构和功能分析结果的可比性。第一层还包括CQA的工艺过程可比性以及CQA分析方法可比性。第二层用于工艺过程控制或其他可能包含在分子表征中的较低优先级的质量属性。当定量评估不切实际或不可能实际操作时,第三层则用于过程监控或其他比较。
第一级:可比性
有两种技术可用于确定可比性:1)等效性检验和2)K西格玛比较。最小样本量是三个或更多批次的原研药分子和三个或更多批次的仿制药产品批次。每个原研药和用于比较的仿制药批次的每个分析项目测量三到六次将有助于提高对分析方法误差的理解。在进行可比性研究之前,所有分析方法都应经过确认或者验证。建议在比较中使用相同数量的批次,但这不是必需的。通常需要对比较研究的设计进行样本量和功效分析,以证明本次研究的设计有足够的功效来可靠地检测比较中使用的平均差异。样本均匀性的评估也是可取的,但不是必需的。
第一层:使用等效性检验的可比性
生物仿制药一词表示可比性研究中的蛋白质或药物分子不完全是生物相同的。当人们想要保证均值不会相差太大时,可以使用等效性测试。换句话说,经过研究证明实际上是等价的。为每个被测参数设置阈值差异的验收标准。如果两组之间的差异显著低于实际上限并显著高于实际下限,则认为均值是等效的。通常,一旦定义了验收标准,就会使用TOST来证明等效性。等效性检验应使用SAS/JMP等统计软件得出结论。
设定可比性验收标准
在等效性测试中使用了三组不同的响应参数:1)两侧限度(规格上限[USL]和规格下限[LSL])仅一侧USL或仅一侧LSL,以及没有规格的限制。实际差异应相对于目标、公差或作为设计余量的函数来查看。验收标准应基于风险进行评估;较高的风险应该只允许很小的实际差异,相反,较低的风险应该允许稍大的实际差异。在证明风险时,应评估累计知识、产品经验和临床相关性。以下基于风险的分析结果验收标准如下表1所示,但不是绝对的;然而,它们是一般情况下典型的基于风险的结果验收标准。

表1.三种情况下的响应参数说明。
下图1中显示的示例在原研药和比较CQA的均值之间的实际限制为4。置信区间必须完全在限制内才能被视为具有可比性。必须确定每个等效性检验的样本量和功效,并在可比性报告中报告。如下图1中的示例表明,对于此CQA,它们具有可比性。等效性检验控制样本中的均值差异和变异、样本大小和定义的风险因素。
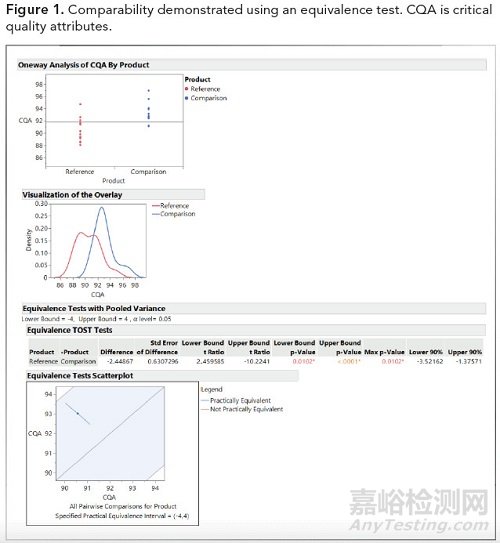
图1.使用等效检验证明的可比性。CQA:关键的质量属性。
K sigma均值测试
另一种显示可比性的可接受方法是基于K西格玛比较。K sigma意味着测试执行起来更简单一些,而且统计上没有那么严格。它采用测试样品减去参考值并除以多个批次和每批次测量值的参考标准偏差之间的平均差。该计算值称为z分数,z分数的绝对值报告为K西格玛。验收标准通常设置为小于或等于参考的1.5K西格玛,以证明可比性。下表2显示了在K sigma中报告的等效性测试中使用的相同数据。这种方法的优点是没有定义严格的规格限,它使用样本均值和变异来确定可比性。
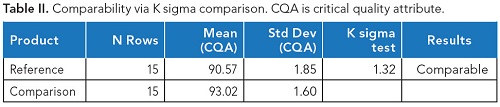
表2.通过K西格玛比较的可比性。CQA:关键的质量属性。
第二级:范围测试
二级范围测试适用于工艺过程控制和那些我们想证明其可比性的不太关键的质量属性,但它们不像一级等效或K σ均值测试中使用的那样严格。正式的风险评估用于确定一级、二级或三级方法。
以下是设置和检查范围测试的过程:
1、使用参考批次仅适用于适当的分布(例如,正态分布、伽玛分布、威布尔分布等)
2、将范围的限制设置为99%(2.576K sigma)或99.73%(3K sigma)(参见下图2)
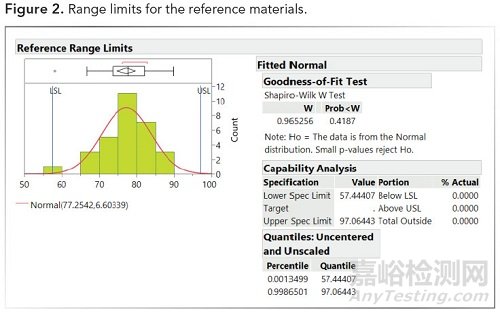
图2.参考结果的范围限制。
3、使用参考限值展示了在参考限值内的比较测量值的百分比(参见下图3)
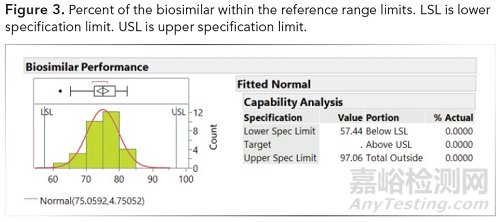
图3.生物仿制药在参考范围内的百分比。LSL:规格下限。USL:规格上限。
4、基于风险,可接受标准可以设置为大于或等于85%、90%或95%。
使用3K sigma从参考材料设定的限值为57.4和97.0,然后应用于生物仿制药以显示可比性。实际百分比(如上图2和图3)等于生物仿制药测量值在参考样品范围内的百分比。
第三级:一般条件
第3级用于在生产过程中简单监控的属性,或用于定量分析不能或不可取的质量属性。第3级仅使用分子结构、生长曲线、传感器测试、混合器中的功率等的图形比较。在大多数情况下,并排图形比较或图形分析的叠加可用于可比性证明。对于第三级,不使用验收标准;然而,重要的是要指出视觉上的可比性,其中区域相似或在比较中已从视觉上检测到差异。如下图4显示了按位置和规模变化对生长曲线的直观比较。
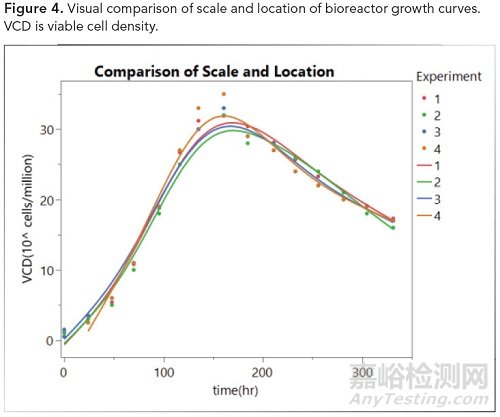
图4.生物反应器生长曲线图的比较。VCD:活细胞密度。
综合比较
强烈建议在进行生物仿制药研究时使用基于风险的方法。例如下表3和表4显示了如何执行比较的示例。

表3.一级评估和汇总表。HMW:高分子量,LMW:低分子量,USL:规格上限,LSL:规格下限。CQA:关键质量属性,TOST:双单边t检验,CI:置信区间。
选择响应,根据它们作为CQA对安全性和有效性的影响和不确定性(风险)的优点,将它们置于适当的层级评估中,说明比较方法,定义验收标准,并得出结论,使组织和报告可比性清晰简洁。还建议使用三级汇总表来汇总结论中评估的属性。

表4.第2级评估和汇总表。VCD:活细胞密度,HCP:宿主细胞蛋白,IPC:处于过程控制中。
总结
证明生物仿制药的可比性是许多制药公司的关键要素。FDA参与制定特定药物的数据和研究要求是证明可比性的关键步骤。一旦确定了研究区域并制定了清晰的属性路线图、抽样计划、样本量和比较方法,开发团队设计和执行可比性协议的效率将大大提高。用于证明可比性的正确组织和适当方法有助于内部和监管当局审查备案和提交报告中的结果值。

来源:药时空