医药洁净室空调系统应能控制空气污染,以保证药品安全性、有效性和质量可控性。尤其作为确保临床使用的无菌药品生产的背景环境,洁净空调系统是无菌药品生产企业的关键的公用系统,药品生产环境得到妥善的设计、建造、调试、运转和维护,则有助于确保产品的质量[1]。中国药品生产质量管理规范(GMP) 规定,应当根据药品品种、生产操作要求及外部环境状况等配置空调净化系统,使生产区有效通风,并有温度、湿度控制和空气净化过滤,保证药品的生产环境符合要求[1]。2022年5月27日国家药监局发布中国GMP 临床实验用药品附录(2022年第43号) 公告,要求2022年7月1日起实施,这是我国首次将临床用药纳入GMP管理范畴,附录内容规定:临床试验用药品的制备和质量控制应当遵循《药品生产质量管理规范》的相关基本原则及数据可靠性要求,最大限度降低制备环节污染、交叉污染、混淆和差错的风险,确保临床试验用药品质量,保障受试者安全。制备临床试验用药品的厂房、设施和设备应当符合《药品生产质量管理规范》及相关附录的基本要求。厂房、设施、设备的确认范围应当基于风险评估确定[2]。
近年来,制药行业的科技及技术在不断进步,不仅在生产设备、实验室设备方面取得了进展,而且在空调系统方面也取得了进展。欧盟GMP附录1“无菌药品生产”中明确提出,无菌药品的生产应符合特殊要求,以尽量降低微生物、微粒和内毒素/热原污染的风险。应考虑以下关键领域应按照GMP指南的相关章节对设施、设备和工艺进行适当的设计、确认和(或) 验证,并在适用的情况下进行持续核实[3]。持续有效的确认活动,保障洁净室洁净级别持续符合产品及工艺需求,对实现向患者提供安全有效药品的目标具有重要影响。
1、洁净室的确认法规要求
各国GMP对药品生产用空调系统均有具体阐述。中国GMP(第四章厂房与设施中规定) : 为降低污染和交叉污染的风险,厂房、生产设施和设备应当根据所生产药品的特性、工艺流程及相应洁净度级别要求合理设计、布局和使用,并符合下列要求。应当根据药品品种、生产操作要求及外部环境状况等配置空调净化系统,使生产区有效通风,并有温度、湿度控制和空气净化过滤,保证药品的生产环境符合要求[2]。美国FDA“211.46通风、空气过滤、空气加热与冷却”[4]、欧盟GMP“3.12”章节,以及WHO GMP通则和PIC /S GMP 指南的第一部分: 医疗产品的基本要求也有类似表述。
查阅各国GMP法规指南对洁净室确认的相关要求,中国GMP无菌附录中仅简单规定了风速标准、单向流验证的要求,也要求“根据洁净度级别和空气净化系统确认的结果及风险评估,确定取样点的位置并进行日常动态监控”[2]。但未明确如何确认及验证。欧盟GMP附录一设置了专门的“洁净室和洁净空气设备的确认”章节,4.32~ 4.32共十项条款对确认形式、项目等,明确阐述了洁净室和洁净空气设备的确认是评估分级的洁净室或洁净空气设备与其预期用途的符合程度的整体过程,应根据所要求的环境特性进行确认,洁净室确认(包括分级) 应与动态环境监测明确区别开来[3]。GBT 25915.1-2021 洁净室及相关受控环境, ISO 14644-1: 2015 洁净室及相关受控环境第一部分: 依据粒子浓度对空气洁净度的分级及GMP确认与验证附录提供了具体的实施指导。
综上可知,不论是美国、欧盟还是中国,都从洁净系统的设计和污染防控、监测等方面提出了系统的要求。中国GMP和FDA均未规定如何进行洁净室确认、确认哪些项目等,仅欧盟GMP附录一有详细要求,确认环境关键要素对维持无菌生产设施符合要求具有重要意义。应在整个设施内实施污染控制策略(CCS) ,以界定所有关键控制点,并评估管理药品质量和安全风险所用的所有控制措施(设计,程序性、技术性和组织性措施) 和监测措施的有效性[3]。
2、洁净室的确认流程
洁净室的确认遵循中国GMP 确认与验证的原则及基本要求,应建立确认与验证的文件和记录,并能以文件和记录证明,并保持持续的验证状态。通常新建厂房设施较为复杂,应满足GMP 要求,也会运用项目管理等方法工具,会基于对产品和工艺的理解,基于产品销售市场的法规要求进行。其核心是界定系统及流程的所有关键控制点,以及系统之间的相互作用,对设施、设备和生产工艺的风险控制措施,并将所有关键控制点和控制措施的有效性进行关联评估。确保环境质量持续满足生产需求。为后续日常监测以及相关控制方法提供依据,确定日常监控频次。
洁净室的确认和验证方法: 通常在现场验证主计划中给出测定方法,以及验证策略。应用于确认的整个生命周期的系统的验证方法。洁净室的确认通常涵盖: 设计确认(DQ) 、安装确认(IQ) 、运行确认(OQ) 、性能确认(PQ) 及再确认等几个阶段。应定期评估持续保持验证状态,当发现趋势出现渐进性变化时,应当评估是否采取纠正措施,评估是否进行必要的再确认。
由ISO14644 中可知,在无菌药品洁净室确认的确认时,主要包括: 已安装的过滤器系统的检漏和完整性测试、气流测试(流速、风量) 及换气次数、自净及恢复时间测试、压差测试、气流方向测试和气流流型研究、温度测定测试、相对湿度测试、悬浮粒子测定、微生物污染监测(浮游菌、沉降菌和表面微生物) 等。洁净室的确认目的是评估洁净室的洁净级别是否满足其预期用途的过程,通常会结合生产需求进行风险评估,测试时需考虑“静态”测试及“动态”测试。在实际测试过程中,设计确认应当证明设计符合用户需求,并有相应的文件。安装和运行确认完成并符合要求后,方可进行性能确认[5]。
根据欧盟附录一4.23用于无菌产品生产的洁净室和洁净空气设备,如单向流单元(UDAFs) 、RABS和隔离器,应根据所要求的环境特性进行确认。每个生产操作要求具有合适的动态下环境洁净水平,以最大程度降低所处理的产品或物料的污染风险。应维持“静态”和“动态”下的适当洁净度水平。可知现阶段对无菌环境的要求更加的贴合实际情况来验证,要求相较更加严格,覆盖更加的广泛,故而在实际操作中的动作影响应多加进行风险评估。
洁净室的确认相对复杂,影响因素较多,结合法规要求及生产实际,应建立洁净区性能确认管控策略,下面举例说明验证确认中可能的问题及需关注要素,见表1。
表1 洁净室确认内容及关注要素
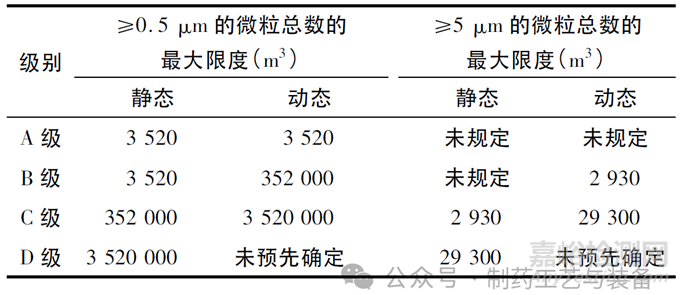
洁净室确认(包括分级) 与动态的日常环境监测应在文件中明确区别并规定。洁净室分级是洁净室确认的一部分,是一种根据洁净室或洁净空气设备的标准通过测定总微粒浓度来评估空气洁净度水平的方法。中国GMP无菌药品附录中,未明确洁净室分级标准。根据欧盟附录一4.27对于洁净室分级,应测定≥0.5 μm 和≥5 μm 的微粒总数。应按照表2中洁净室分级时各级别最大允许的总微粒浓度中规定的限度,在静态和模拟操作中都进行测定。
表2 洁净室分级与各级别最大允许的总微粒浓度
由表2可知,对A级≥5 μm 的微粒总数与B级静态的≥5 μm 的微粒总数要求更新未规定,实际控制标准及要求无变化,仅根据在实践过程中,尤其是针对新建或改扩建厂房设施,应当对暴露的产品及部件足够保护的状况下,监测动态的实际意义不大,基于风险的简化了验证的流程。在洁净分级时,应在静态和模拟操作中都进行测定。
由表3可知,对A级最大允许的微生物污染水平的要求更新为无生长,该描述更加贴合实际,更科学合理,也更能看出监管机构不认可平均后<1 的说法及做法。对未来的日常监测及无菌理念有一定的正向影响。
表3 确认过程中最大允许的微生物污染水平
3、洁净室的确认实施问题思考
洁净室的确认是一项综合、系统的、复杂的工程,会受到多种因素影响,与厂房设施的设计、控制、监测等多维度、多因素有关,如洁净室的确认可能受各相关因素或系统影响[3]。在洁净室确认实践中,经常会发生不符合或偏差,应在确认方案中明确偏差处理流程,确保所有区域符合要求。常见问题及解决方法,见表4。
表4 常见问题及解决方法
在欧盟附录一发布后,气流流型研究成为了业内讨论较多的话题。(1) A 级单向流风速测定: 在工作区域应满足0.36 ~ 0.54 m/s(指导值) 均匀风速,除非在CCS 中另有科学论证,气流可视化研究应与风速测定相关联[3]; (2) 气流流型拍摄: 现阶段对气流流型研究的效果及目视观察的形状要求更高,需在符合实际情况的动态操作下进行,且气流流型清晰可见,在拍摄途中可准备黑色幕布以保证清晰度,同时需注意拍摄需有设备与拍摄目标的铭牌或编号,为确保数据可靠性有时会考虑采用三维视频拍摄。
4、小结
有效的无菌药品洁净室确认是质量源于设计、质量源于控制策略的最佳实践,洁净室确认不是一次性确认,应基于法规要求及日常监测趋势预测变化,持续有效的确认是保障药品无菌关键质量属性的前提及必要条件。虽欧盟、FDA和我国对无菌药品及洁净室确认的描述不尽相同,但从其核心是以生产工艺需求的评估、设计、确认、监测和控制为基础,基于对监测数据所采集的信息及相关的特定GMP要求符合性的审核,更全面、更准确进行产品无菌保证能力的评价。基于风险管理洁净室全生命周期确认实施策略,为后续生产制造确定污染防控策略,建立失败的风险发现机制。其目的是增加上市产品的无菌保障水平,确保患者用药安全,尤其针对避免处于临床用药期间的适用人群、受试者的风险变得更为重要。
参考文献
[1] 冀红. 论洁净区( 室) 动态监测中在线监测系统的必要性[J]. 临床医药文献电子杂志, 2018,5(30) : 184-185.
[2] 中华人民共和国卫生部. 药品生产和质量管理规范( 2010 年修订) [S]. 卫生部令第79 号.
[3] European Commission. EU GMP Annex 1: Manufacture of Sterile Medicinal Products [EB /OL]. ( 2022-08-22) [2023-01-10]. https: / /health. ec. europa. eu /system/files /2022-08 /20220825 _ gmp-an1 _en_0. pdf.
[4] CFR 联邦法规: 第21 篇“食品和药品”第211 部分制剂成品的现行生产质量管理规范.
[5]ISO14644-1Cleanrooms and associated controlled environments Part1: Classification of air cleanliness by particle concentration.

来源:临床合理用药
关键词:
无菌药品洁净室