摘要:分析兰索拉唑肠溶制剂抽检中溶出度异常产品的原因并提出改进建议。按照法定标准对抽检制剂进行检验,一批产品的溶出度低于限度,其他项目均符合规定。考虑到本品对酸、碱不稳定,结合对生产企业飞行检查的结论,设计实验考察该批次产品溶出度异常的原因,包括高温高湿环境对溶出度、含量和有关物质的影响;2h 抗酸实验对溶出度的影响;利用激光红外成像系统进行片芯成像及包衣层厚度测定;有关物质色谱条件优化;利用轨道阱高分辨质谱进行杂质来源分析等。研究发现,流通环节贮存不当和产品包衣工艺差是引起该批次溶出度低的原因。流通环节可能出现的高温高湿环境引起制剂中崩解剂功效下降,主药难以完全释放;包衣液不能均匀包裹于片芯,引起隔离层薄且厚度不一,继而影响其保护主药防止酸降解的效果;两者共同造成该批次产品溶出度偏低。兰索拉唑肠溶制剂总体质量较好,但部分企业处方和工艺需要优化;流通环节需要严格按照规定控制温湿度。
兰索拉唑是日本武田制药株式会社研制的质子泵抑制药,其抑制胃酸分泌作用及抗幽门螺旋杆菌作用较强,在治疗消化性溃疡疾病方面具有显著疗效。从国家药品不良反应监测系统数据库中查询到兰索拉唑肠溶片和肠溶胶囊存在皮疹、瘙痒、肝功能异常、过敏性休克、血小板减少等不良反应。目前兰索拉唑肠溶片收载于《中华人民共和国药典》(以下简称《中国药典》)和《英国药典》,兰索拉唑肠溶胶囊收载于《中国药典》、《美国药典》、《日本药典》和《英国药典》。天津武田和日本武田的兰索拉唑肠溶胶囊均为国家药品监督管理局公布的参比制剂,本研究以天津武田生产的肠溶胶囊为原研制剂进行相关研究工作。
根据法定标准对在全国范围内抽取的兰索拉唑肠溶片和肠溶胶囊进行检验,发现H企业一批样品在pH6.8 磷酸盐缓冲液中的溶出度低于限度,其他项目均符合标准规定。根据能量守恒原则,推测企业存在低限投料的可能,或者有其他降解产物在法定检验中未检出。结合H局对H企业飞行检查的结论,在探索性研究过程中,采用激光红外成像技术考察该企业不同批次产品片芯及包衣喷涂的均匀性,利用正交分析技术优化现行药典方法中有关物质的色谱条件,结合LC-Orbitrap/HRMS 及多中心切割2D-LC-HRMS 技术,对本品杂质来源进行系统归属,最终认为流通环节贮存不当和产品包衣工艺差是导致该批次产品在pH6.8磷酸盐缓冲液中溶出度偏低的原因,本研究工作可以较好地推进本品仿制药一致性评价的进程。
1、 材 料
1.1 药品与试剂
兰索拉唑(批号100709-201705,含量99.6%)、兰索拉唑杂质Ⅰ(批号510046-201401)、兰索拉唑杂质Ⅱ(批号 510047-201401)、兰索拉唑杂质Ⅲ(批号510048-201401)、兰索拉唑杂质Ⅳ(批号510049-201401)(中国食品药品检定研究院);兰索拉唑杂质C(批号S17041902,含量99.94%)、兰索拉唑杂质F(批号S17110601,含量99.3%)(江苏奥赛康药业有限公司);兰索拉唑肠溶片和兰索拉唑肠溶胶囊为抽样样品。乙腈、三乙胺(德国默克公司);甲酸、甲醇(美国赛默飞世尔公司);其他试剂均为市售分析纯。
1.2 仪 器
LC-20AB 高效液相色谱仪、UV-2600 紫外分光光度计(日本岛津公司);1260 Infinity Ⅱ高效液相色谱仪、708-DS 溶出仪、850-DS 溶出自动取样器、8700 LDIR 激光红外成像系统(美国安捷伦公司);Ultimate 3000-QExactive Orbitrap 液质联用(美国赛默飞世尔公司)。
2、 方 法
2.1 法定检验方法
本次抽样的兰索拉唑肠溶片法定标准主要为《中国药典》2015年版二部;兰索拉唑肠溶胶囊法定标准主要为《中国药典》2015年版第一增补本,少数样品执行国家药品监督管理局标准。
2.2 探索性研究方法
对兰索拉唑肠溶片溶出度异常产品的原因进行了不同层面的探究,包括高温高湿环境对该品种溶出度、含量和有关物质结果的影响;2 h抗酸实验对溶出度结果的影响;利用激光红外成像系统对溶出度异常批次样品进行片芯成像以及包衣层厚度的测定;优化有关物质色谱条件以分离出更多杂质;对该批次产品中的杂质进行来源分析等。
3、 法定检验结果与讨论
在法定检验中发现H企业一批产品溶出度低于限度要求,该批次为本研究的重点。通过调研,H企业生产的190301批次兰索拉唑肠溶片于2019年4月26日生产入库,后分别于2019年5月14日、5月26日分两次发往J企业,2019 年 6 月28日由J企业发往Y公司,再由Y公司分销1件(300 盒)产品至 S 公司,即本次检验溶出度异常产品的抽样场所。
本次法检报告公示之后,H 企业迅速展开该批次产品的召回工作,并将从全国各地召回的产品送至 H 省院进行检验,结果见表 1,可知该批次从其他省市召回的产品溶出度项目均合格,从 S 药业召回的产品溶出度项目仍然低于限度(限度为 80%),结果比本次检测结果更低。H局对 H 企业进行了飞行检查,调查其批生产记录等资料,均未发现重大问题,初步认为市场贮存不当是导致产品不合格的主要原因,市场贮存中未达到规定的遮光,密封,置阴凉(不超过 20 ℃)干燥处保存的要求,因而对溶出度的结果造成了影响。
4、 探索性研究结果与讨论
4.1 高温高湿环境条件对产品关键质量属性的影响
为了确证 H 局的调查结果,本研究考察了高温高湿条件对产品关键质量属性的影响,将原研制剂和 H 企业的一批次产品在高温高湿即温度 40 ℃、相对湿度 75%(RH 75%)的贮存条件下放置5天后,采用法定检验的标准检查溶出度、含量和有关物质,并与放置在规定的阴凉(不超过20 ℃)干燥贮存条件下的同批次样品检验结果进行比较。结果见表2—4。



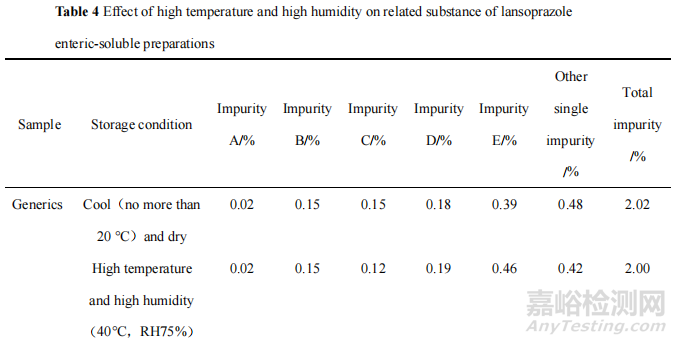

数据表明,在高温(40 ℃)和高湿(RH 75%)环境中放置后,兰索拉唑肠溶片溶出度明显降低,含量和有关物质却无显著变化。推测在高温高湿环境中,水分会引起片芯中的崩解剂膨胀,因而在溶出度试验中,崩解剂功效下降,造成溶出度结果偏低。这进一步证明市场贮存不当可能是导致该产品溶出度异常的原因之一。
4.2 产品包衣层工艺对溶出度结果的影响
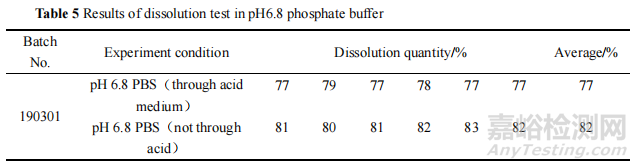
对该批次样品在酸介质中的溶出量进行检测,发现其在酸介质中经 2 h 始终未溶出,性状也无明显变化。将该批次肠溶片直接置于 pH 6.8 磷酸盐缓冲液中进行溶出度试验,结果该批次样品的溶出度结果符合标准规定。表明该批次样品虽然没有在酸介质中溶出,但是法定检验方法中前期抗酸 2 h 的步骤对最终的溶出度结果有影响,提示本品包衣层存在缺陷。结果见表 5。
通过 8700 LDIR 激光红外成像系统对该批次样品切片后的片芯进行成像,并与辅料单组分成像结果进行比较。结果显示片芯中含有兰索拉唑、甘露醇、低取代羟丙甲纤维素、碳酸氢钠和羧甲淀粉钠,与厂家提供的处方一致。并将激光红外成像结果计算得到的各组分平均面积占比与厂家提供的处方含量进行对比,两者有轻微差异,这可能由于得到的切片不是最中间面导致的,说明企业在生产过程中确实按处方投料。
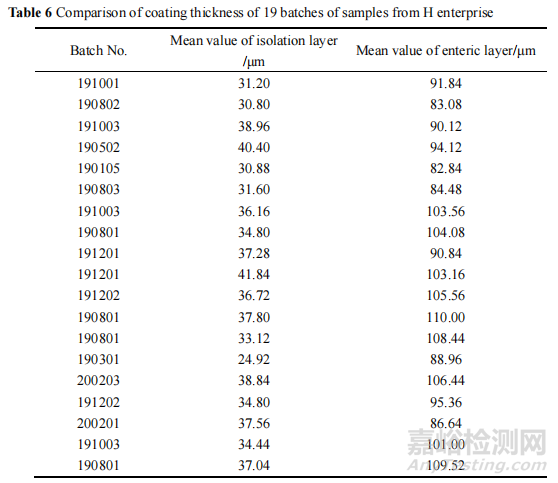
再通过 8700 LDIR 激光红外成像系统,利用隔离层和肠溶层特征峰的不同对包衣进行成像,成像后利用软件标尺测量隔离层包衣与肠溶层包衣厚度的方法,测定本次抽到的 H企业不同批次样品的包衣厚度。
结果显示该批次(190301)的隔离层是所有样品中最薄的,平均厚度仅为24.92 μm,其他批次样品隔离层平均厚度在 30.80~41.84 μm 之间。各批次样品的肠溶层平均厚度在82.84~110.00μm 之间,测定结果见表 6。
此外,在成像图发现 190301 批样品的隔离层厚度不均一,部分偏薄,部分偏厚,且个别地方存在隔离层断层现象,其厚度及完整性均较差。调研得知,隔离层在该制剂中具有两个主要作用:一是隔离主药与肠溶衣层防止直接接触引起主药降解;二是修饰片芯边角,提高肠溶性能。推测隔离层的厚薄不一易引起主药遇酸降解,导致溶出度下降。另一方面,由于隔离层薄,溶出度试验中的盐酸溶液会渗透进片芯,造成片芯中的崩解剂迅速膨胀,再转移至 pH 6.8 磷酸盐缓冲液后,崩解剂的崩解效果大打折扣,导致主药释放不完全,引起溶出度下降。综上所述,隔离层薄引起酸降解和崩解剂效果差可能是导致该产品溶出度不合格的另一个因素。
4.3 利用多中心切割 2D-LC-HRMS 和 LC-Orbitrap/HRMS 技术对溶出度不合格批次降解情况的探索
根据上述研究中的推测,该批次产品的主药应该有降解,且为酸降解,非高温高湿降解。但在法检中,该批次的含量和有关物质均符合标准规定。最大单杂含量为 0.4%(限度为0.5%),总杂含量为 0.8%(限度为 2.0%),为了进一步探究该产品的降解情况,我们做了以下探索性研究。
4.3.1 基于 2D-LC-HRMS 对兰索拉唑肠溶片中法检发现的主要杂质进行研究采用ThermoUltimate 液相与 Thermo Q-ExativeOrbitrap 质谱联用技术,一维色谱条件同《中国药典》有关物质项下方法,二维色谱条件采用 0.1%甲酸水-乙腈为流动相,梯度洗脱。按照《中国药典》规定,相对保留时间 0.25 之前的色谱峰忽略不计,结果显示,总计得到 9 个杂质,编号为 1~9,其中峰 1 在该色谱条件下的紫外谱图中未能完全分离,但质谱提取离子流图可以实现有效分离,故标记为杂质 1 和 2,详见图 1。通过与已知杂质的保留时间和质谱数据比较,归属了 5 个色谱峰为已有对照品的已知杂质,分别为欧洲药典杂质 D、E、A、B、C,在图中色谱峰编号依次为 1、2、6、7、9,剩下的未知杂质为 3、4、5、8。
4.3.2 有关物质色谱条件的优化根据上述研究结果可知,《中国药典》的方法无法分离杂质 D和 E,且规定相对保留时间 0.25 之前的色谱峰忽略不计,对本次抽检的 H 企业所有样品的色谱图进行对比,发现相对保留时间 0.25 之前的色谱峰并完全不一致,其中 190301 批的色谱峰明显大于其他批次,推测有部分降解产物随辅料峰一并在相对保留时间0.25之前流出。因此我们通过正交设计优化了有关物质测定方法,并对溶出度异常样品进行了测定。结果显示,优化后的方法将《中国药典》中未能分开的杂质 D 和杂质 E 分开了,而且无论是检出杂质个数还是杂质总量都明显大于《中国药典》方法。溶出度异常批次的总杂,从 0.84%变成 4.84%,最大单杂从 0.35%变成2.14%,色谱图见图 3,结果证实有降解杂质未被发现。
4.3.3 基于 LC-Orbitrap/HRMS 对二维质谱系统中未检出杂质进行研究在“4.3.2”优化后的有关物质方法基础上,采用高效液相色谱——高分辨轨道阱质谱联用(LC-Orbitrap/HRMS)技术,建立了兼容质谱检测器的检测方法。一共检出杂质 14 个,其中 5 个为欧洲药典杂质D、E、A、B、C;9 个为未知杂质,未知杂质中 4 个与利用 2D-LC-HRMS 技术检出的结果一致,5 个为该条件下新检出杂质(命名为杂质 10-14)。根据兰索拉唑原料强制降解实验结果,原料药合成工艺以及文献资料[1-5],对杂质来源进行分析。发现 H 企业溶出度不合格产品中新出现的最大单杂(杂质 11)为酸降解杂质,详见图 2 和图 3。该结果进一步证实了我们对隔离层薄引起酸降解的推测。
5、 小 结
本次研究采用激光红外成像技术、多中心切割 2D-LC-HRMS 等技术,结合异常批次样品的自身特性以及调研结果,从不同维度设计试验,解析溶出度偏低的原因,包括外部环境因素以及产品自身问题。对于研究中得出的导致隔离层薄的原因,是隔离层喷雾工艺参数设置不佳或是隔离层材料选择不优造成,目前还未能得出结论。由于兰索拉唑肠溶片目前未有通过仿制药质量和疗效一致性评价的产品,可见该品种在研发中对辅料和工艺的筛选是具有挑战和技术难度的。
因目前的检验方法未能很好地分离和检出部分杂质,后通过正交设计优化的有关物质方法证实了该产品主药有酸降解的现象,更加互补验证了该批次产品隔离层薄,引起了主药降解的情况。虽然本次研究发现的问题无致命安全隐患,但也存在一定的风险,因此建议提高该品种标准,主要是有关物质方法,上报药典会;同时建议经营企业在药品流通过程中严格按照药品规定的储存条件进行;建议生产企业对影响本产品质量的因素如投料量、包衣工艺、包衣材料选择等进一步深入研究,严把原料、生产工艺关,特别是改进不合理的工艺参数。
产品质量是仿制药一致性评价的核心,仿制药的质量应与原研药在成分、药效、安全性、稳定性等方面保持一致,以确保患者使用仿制药能够获得与原研药相同的疗效和安全性。为了保证药品质量和稳定性,仿制药的生产工艺应与原研药相似,否则可能因为工艺差异影响药物疗效和安全。本研究采用了多种技术开展研究工作,可以较好地推进本品仿制药质量和疗效一致性评价的进程。多中心切割 2D-LC-HRMS 技术在药物杂质谱研究中已经得到了广泛应用,在评价仿制药质量中正发挥着重要作用。另一项关键技术——激光红外成像技术可以协助进行处方分析,对不同来源的辅料也具有一定区分能力,其在仿制药一致性评价中的应用将大为可观。采用多种技术从不同维度对仿制药的产品质量、处方工艺等进行深入研究,可以全面分析对比仿制药和原研药的质量属性,找到影响产品质量的主要参数,改进仿制药生产工艺。
参考文献:

来源:Internet
关键词:
仿制药
一致性评价