目前溶出方法开发及验证方面,USP<1092>讲解的较为详细,本文中的验证一般要求,主要是参考了目前USP 2023版本的内容。在国内的指导原则,也明确指出了仿制药溶出曲线检测应该进行必要的验证,如《普通口服固体制剂溶出曲线测定与比较指导原则》,指出“溶出试验方法应能客观反映制剂特点、具有适当的灵敏度和区分力”,“方法建立后应进行必要的验证,如:准确度、精密度、专属性、线性、范围和耐用性等”;《化学药品仿制药口服固体制剂质量和疗效一致性评价申报资料要求(试行)》指出“体外溶出曲线测定方法(含多个溶出介质)建立后,按照中国药典2015版四部附录〈9101〉药品质量标准分析方法验证指导原则进行必要的方法学验证,如:准确度、精密度、专属性、线性、范围和耐用性等。”。
溶出曲线的验证与溶出度相比,主要区别在于范围。溶出曲线以第一个溶出点为低限,最终溶出量为高限;溶出度以标准溶出量为中间点进行研究,相比溶出度,溶出曲线的研究范围要宽一些。
以溶出度方法学验证为列,以下是我对于溶出方法学验证的个人总结。
一、参考依据
溶出方法学较一般的含量方法学验证,除了需要验证分析(检测)方法,还需要验证溶出(溶出过程)方法。对于完整的溶出度方法学验证,主要参照中国药典、美国药典以及ICH文件,如下:
● 中国药典2020版,9101分析方法验证指导原则。溶出度分析方法验证指标包括:专属性、准确度、精密度、线性和范围、耐用性。
● USP〈1225〉 Validation of Compendial Procedures。
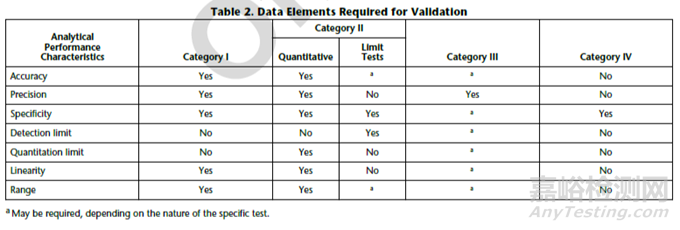
CATEGORY III:Analytical procedures for determination of performance characteristics (e.g., dissolution, drug release, and others).
● USP〈1092〉 The Dissolution Procedure Development And Validation。分析方法验证指标包含专属性、线性和范围、准确度、精密度、耐用性;溶出方法验证指标包含精密度、耐用性。溶出方法学验证使用具有代表性的均一样品。
● 【中文】ICH Q2(R2):分析方法验证(草案)(2023.11.01采纳,2024年CDE发布的中文翻译稿)。对其他定量测定,做出了说明,“如果工作范围接近技术的检测限度或定量限度,则其他定量测定可遵循杂质检查方案,否则建议遵循含量测定方案”。同时将速释制剂的溶出度方法学验证作为其中的示例,溶出步骤的验证指标为精密度、耐用性;分析方法的验证指标为专属性/选择性、线性和范围、准确度、精密度、耐用性,其中范围下限是根据具体情况而定,如果工作范围下限接近定量限,是需要验证定量限的。
二、方法学验证过程
(一)专属性
目的证明溶出介质与辅料对样品检测无干扰;验证过程及可接受标准来源 USP<1092>。
溶液配制
空白溶液:取溶出介质,按照供试品的处理方式进行处理(如过滤等)。
空白辅料溶液:取空白辅料混合样品(含包衣粉、胶囊壳)适量(按处方比例及介质体积换算),在37℃的条件下,在溶出介质中溶解或分散,按照供试品的处理方式进行处理(如过滤等)。
供试品溶液:取空白辅料混合样品适量(按处方比例及介质体积换算;缓控释制剂,更建议采用空白制剂的方式进行),在37℃的条件下,加入溶出介质溶解或分散,按处方比例加入适量原料药,使完全溶解(为确保溶解完全,也可按处方比例加入适量的原料药贮备液),按照供试品的处理方式进行处理(过滤等)。
其他:当样品在介质中不稳定,存在显著降解,需要增加降解物的干扰实验。
使用紫外法时:空白溶液测定,扣除比色皿+空气空白;空白辅料溶液测定,扣除溶出介质空白;空白溶液测定时,扣除辅料空白。
上述供试品溶液,也可以用对照品溶液代替,按照以下计算公式计算干扰:
Result = (AP/AS) × CS × (V/L) × 100
AP=空白的吸光度值或响应值
AS=供试品溶液的吸光度值或响应值
CS=供试品溶液的主成分浓度(mg/ml)
V=介质体积
L=标示量(mg)
可接受标准
HPLC法:溶出介质、辅料均不干扰主峰检出;主峰纯度应符合要求。
UV法:按上述计算方式,溶出介质干扰应不超过1%;空白辅料干扰不得过2%。
(二)定量限/检测限
应根据最小溶出量、释放量决定是否需要进行检测限/定量限考察。一般,缓控释制剂以及迟释制剂不释放或几乎不释放的溶出介质中考察;速释制剂无需进行该研究。需不需要做定量限可参照下述线性与范围项下的描述。
HPLC法:采用对照品,用相应的溶出介质逐步稀释进样。LOD信噪比2:1或3:1;LOQ信噪比10:1。
UV法:按照公式计算。
LOD = 3.33δ /S ;LOQ = 10δ /S。
式中,δ:响应值的偏差;S : 标准曲线的斜率。
δ可以通过下列方法测得:① 测定空白值的标准偏差;②标准曲线的剩余标准偏差或是截距的标准偏差。
可接受标准
定量限定结果应符合准确度和精密度要求。连续进样6针定量限溶液,主峰峰面积RSD≤10%。(也可根据中国药典9101指导原则,表3 样品中待测定成分的含量与精密度可接受范围关系,制定相应的RSD值要求)
(三)线性和范围
线性溶液配制:可用一贮备液经溶出介质精密稀释,或分别精密称样,制备一系列被测物质浓度系列进行测定,至少制备5个浓度。以测得的响应信号作为被测物浓度的函数作图,观察是否呈线性,用最小二乘法进行线性回归。
应注意,如果对照品/原料药不易溶解,可考虑添加有机溶剂溶解制备贮备液,但最终线性溶液中有机溶剂的使用体积应小于5%。
可接受标准
r≥0.99(r2≥0.98,USP)
范围:一般,溶出度限度的+20%,如规定限度范围,线性范围则为下限-20%至上限+20%。线性范围下限接近0%,应进行检测限/定量限的考察,线性范围的下限应为定量限。详细可参照下述各指导原则等,考察线性范围。
(四)回收率
溶液配制
对照品溶液配制:按照拟定方法进行配制。
供试品溶液配制:取所有混合辅料(溶出检测方法下的供试品100%浓度水平,应包含包衣粉、胶囊壳等),在37℃的条件下,加入对照品或已知含量的原料药配制3种浓度,每种浓度3份样品,加入溶出介质溶解,按拟定的供试品的处理方式进行处理(如过滤等)。加入对照品或原料药的方式,可以为逐份准确称量,也可以先配制贮备液,精密量取适量体积的方式进行。如使用有机溶剂制备贮备液,贮备液总有机溶剂体积含量应不超过5%。
3种浓度一般分别为线性范围的低限、限度浓度(100%浓度)、线性范围的高限
计算公式:回收率=测得量/加入量×100%
可接受标准
回收率范围95.0%~105%(标准来源USP<1092>)
其他情况:对于特殊情况,如USP<711>中所述,第一阶段释放量不超过10%,第二阶段释放量不超过25%等,做回收率试验具有挑战性,可参照USP<1092>/5.Validating/5.3 Accuracy/Recovery项下内容。
(五)精密度
精密度试验分为分析方法的精密度试验与溶出方法的精密度试验。可参照ICH Q2(R2)、USP<1092>中的内容。ICH Q2中区分了溶出方法的精密度试验、分析方法的精密度试验,溶出方法的精密度试验使用一批具有代表性的制剂考察,分析方法的精密度考察使用1片制剂进行考察,备注中说明溶出方法的精密度研究将允许对制剂和分析的变异性进行综合评估 ;USP中中间精密度对溶出方法和分析方法进行综合评估,未给出具体的可接受标准。
精密度(分析方法)
重复性样品配制
对照品溶液:按照拟定方法进行配制。
供试品溶液配制:
第一种方法(见ICH Q2(R2)):使用1个/片/粒制剂,用溶出介质完全溶解后,按照供试品处理方式(如过滤等)配制6个样品;
第二种方法:与100%回收率样品配制方法一致,配制6份样品。
重复性可接受标准(分析方法)
同一对照品溶液连续进样6针(HPLC法)/测定6次(紫外分光光度法),6次对照品溶液的响应值(峰面积或吸光度值),RSD<1%;测得6份供试品溶液的溶出量,RSD<2%(USP2023 <1092>)。
中间精密度样品配制
不同试验员、不同日期、不同液相/紫外分光光度计,按照上述重复性操作过程,配制溶出介质及样品。
中间体精密度可接受标准(分析方法)
12份供试品溶液的溶出量,RSD<2%。
精密度(溶出方法)
重复性样品配制
对照品溶液:按照拟定方法进行配制。
供试品溶液配制:使用有代表性的均一产品,以溶出度检测方法进行研究。取6片,使用溶出仪配制的溶出度样品并依据拟定方法检测。
注:如果没有合适的产品,可以使用原料药+辅料的形式,进行检测。
重复性可接受标准(溶出方法)
溶出度RSD不超过10%,溶出量较低的情况可放宽至20%
中间精密度样品配制
不同试验员、不同日期、不同溶出仪、不同液相/紫外分光光度计,按照上述重复性操作过程,配制溶出介质及样品。
中间精密度可接受标准(溶出方法)
12份溶出度RSD不超过10%,溶出量较低的情况可放宽至20%;
溶出度<85%的情况,两次检测样品的溶出度结果平均值的差值的绝对值不超过10%,溶出度不小于85%的情况,两次检测样品的溶出结果的平均值差值的绝对值不超过5%。
说明:中间精密度溶出度差值的绝对值要求的来源为USP<1092>,5.4.2 INTERMEDIATE PRECISION/RUGGEDNESS;溶出度RSD值的要求参考USP<1092>,2. METHOD DEVELOPMENT,原文“One guidance defines dissolution results as highly variable if the relative standard deviation (RSD) is more than 20% at time points of 10 min or less and more than 10% at later time points for 12 dosage units tested”,如果RSD超出20%或10%认为是高变异。这里参考的指南指的是USP<1092>参考文献(15),详细可查看USP<1092>。
(六)耐用性
耐用性同精密度一致,需要验证分析方法的耐用性和溶出方法的耐用性。
耐用性(分析方法)
样品配制
对照品溶液:按照拟定方法。
供试品溶液:1份;使用制剂溶解或100%回收率的做法。
同一般分析方法学验证相同,液相色谱法中典型的变动因素有流动相的组成和pH值、不同品牌或不同批号的同类型色谱柱、柱温、流速等。USP<1225>提到了改变检测波长,目前国内是不改变波长的;对于紫外分光光度计法,一项变动因素是检测波长。关于USP中描述原文见下方:
Robustness of analytical finish is referenced in <1225>. HPLC analysis parameters may include mobile phase composition (percentage organic, buffer concentration, pH), flow rate, wavelength, column temperature, and multiple columns (of the same type). For spectrophotometric analysis, the wavelength may be varied.
可接受标准(分析方法)
改变色谱条件后,与标准条件相比,应在98%~102%范围内;每种条件下,系统适用性应符合要求。
耐用性(溶出方法)
ICHQ2:选择溶出方法参数的依据,例如,溶出介质组成、缓冲剂或表面活性剂浓度、沉降片的使用、pH值、脱气、容积、搅拌速率、采样时间
USP<1092>
The parameters may include medium composition (e.g., buffer or surfactant concentration, pH, deaeration), volume, agitation rate, sampling time, and temperature. Statistical analysis of the data generated will help determine the extent to which the parameters must be controlled in the method.
参数可包括溶出介质组成(如缓冲液或表面活性剂浓度、pH 值、脱氧)、体积、搅拌速率、取样时间和温度。
对于溶出方法的耐用性研究,可作为方法开发过程中的内容。ICH与USP两者基本一致,可作为参考。
样品配制
对照品溶液:按照拟定方法。
供试品溶液:按照上述条件,每种条件从质量均一的制剂中取6片制剂考察,计算溶出度。
可接受标准
每种条件下,6片制剂的溶出度,RSD<2%。
改变溶出条件后,与标准溶出条件下所测溶出度平均值相比,溶出度<85%的情况,差值的绝对值不超过10%,溶出度不小于85%的情况,差值的绝对值不超过5%。
(七)溶液稳定性
考察供试品溶液的稳定性,分别考察37℃、放置温度(如室温、冷藏等条件)两种情况,考察对照品溶液、系统适用性溶液放置条件下的稳定性。考察时间考虑多批次检样、样品配制过程等情况。
可接受标准
与0时样品响应值相比,应在98%~102%范围内。
三、溶出度、溶出曲线方法学验证项目总结

来源:注册圈
关键词:
仿制药
口服固体制剂
溶出方法学验证