
嘉峪检测网 2024-04-20 20:15
导读:本文对医疗机构建设多组学自制试剂实验室的必要性、建设实践和运行风险等进行阐述。
摘 要 / Abstract
实验室自制试剂(laboratory-developed test,LDT)是指由实验室内部开发使用、不需要经过商业注册的体外诊断试剂(in vitro diagnostic medical devices,IVDs)。自制试剂涉及技术广泛,如高通量测序技术、质谱技术、流式细胞分析技术等,能够结合基因组学、转录组学、蛋白质组学、代谢组学等多种组学技术,成为研究遗传病生理病理机制的重要技术方法。中日友好医院被指定为我国10 家自行研制使用体外诊断试剂的试点医疗机构之一,率先开展多组学自制试剂实验室建设。本文对医疗机构建设多组学自制试剂实验室的必要性、建设实践和运行风险等进行阐述。
Objective: Laboratory-developed test (LDT) refers to in vitro diagnostic medical devices (IVDs) developed and used internally within the laboratory, requiring no commercial registration. LDTs involve a wide range of technologies such as highthroughput sequencing, mass spectrometry, flow cytometry, and bring together various omics technologies such as genomics, transcriptomics, proteomics, and metabolomics. They have become an important technical approach for understanding the pathophysiological mechanisms of genetic diseases. China-Japan Friendship Hospital, designated as one of the ten pilot medical institutions for LDTs in China, has taken the lead in constructing a laboratory for multi-omics LDTs. This article elaborates on the necessity, practices, and operational risks of building a laboratory for multi-omics LDTs in a medical institution.
关 键 词 / Key words
医疗机构;多组学;实验室自制试剂;体外诊断试剂;监管
medical institution; multi-omics; laboratory-developed test; in vitro diagnostic medical devices; regulation
欧美等国家和地区的医学中心或临床实验室将实验室自制试剂(laboratory-developed test,LDT)作为商业注册试剂(CE-certified IVDs,CE-IVDs)的补充,用于解决临床紧急需求, 对医疗的发展具有重要意义。在我国,根据新修订《医疗器械监督管理条例》第五十三条, 支持有条件的医疗机构自行研制使用体外诊断试剂( 以下简称自制试剂)实践[1]。由于我国自制试剂的制备、应用及其监管体系与国外现行体系存在差异, 国家药品监督管理局会同国家卫生健康委员会发布《关于开展医疗机构自行研制使用体外诊断试剂试点工作的通知》,将中日友好医院等10 家医疗机构定为试点单位, 以探索我国自制试剂的应用和管理模式。中日友好医院结合学科发展特点率先开展多组学自制试剂实验室建设, 为监管部门和其他医疗机构提供“先行先试”经验。本文对医疗机构建设多组学自制试剂实验室的必要性、建设实践和运行风险进行阐述。
1、医疗机构建立多组学自制试剂实验室的必要性
1.1 自制试剂的定义
国际上,一般使用美国食品药品监督管理局(FDA)对于自制试剂的定义:医学检验实验室自行研发、验证和使用的检测方法,仅在研发实验室内部使用,不作为商品出售的IVDs[2]。欧美等国家和地区的实验室检测通常同时使用CE-IVDs、科研试剂(research use only,RUO)和实验室内部试剂(in-house IVDs,IH-IVDs)。一般认为IH-IVDs 等同于实验室自制试剂,但是也有部分国家和地区将实验过程中对CE-IVDs 进行重大改变或将RUO 应用于临床检测归属于自制试剂范畴[2-3]。
1.2 国内外自制试剂开展情况
国内外自制试剂的开展情况不一致。调查显示,欧盟95% 的实验室检测通过CE-IVDs 完成,剩余部分则需要通过自制试剂完成[4]。在美国,从临床实验室出现伊始,自制试剂就开始应用[1]。据统计,美国LabCorp 检测机构开展的自制试剂项目达5000 多项[5],形成了FDA 与医疗保险和医疗补助服务中心(CMS)的“放”和“管”监管体系[6]。欧洲和日本的实验室也开展了自制试剂项目,但是没有明确的监管定义和与此相关联的医疗费用支付报销制度。其中,日本大约有40 个自制试剂的检测费用可以由医保支付[7] ;德国SYNLAB 检测机构开展的自制试剂项目多达5000 多项[5]。
我国在2021 年之前,由于支持自制试剂发展的相关政策尚未出台, 该领域相较于欧美等国家和地区起步较晚。文献统计显示, 我国三级甲等医院开展的自制试剂项目在500 项左右, 无论是数量还是种类都与欧美等国家和地区相距甚远,且大多仅用于临床研究, 无法满足临床诊疗日趋增长的个体化与精准化需求[5,7]。
1.3 自制试剂监管
在美国, 只有通过美国《临床实验室改进修正案》(Clinical Laboratory Improvement Amendments,CLIA) 认证的实验室才可以进行自制试剂检测,并且这些实验室必须拥有符合标准的质量保障体系、设备设施并配备相应资质的人员等。CMS负责监管自制试剂业务,并制定了临床实验室人员专业性、质控能力、产品验证和实验记录等方面的相关标准。然而,在当前的标准下,自制试剂的准确性没有得到充分监督,这可能会导致巨大的安全隐患,特别是对于高风险的检测,其结果的准确性将影响临床决策,甚至可能危及患者安全。因此,需要加强自制试剂监管框架,从而确保临床检测安全有效进行。自2020 年起,美国《验证准确的前沿体外临床检测发展法案》(Verifying Accurate Leading-edge IVCT Development Act,VALID)多次被提案至美国国会,旨在授予FDA 审查和批准体外临床检测的权力,但均被否决。2023 年9 月, 美国FDA发布的监管新规征求意见稿指出,即便检测试剂是实验室生产的,也将被按照IVDs 进行监管。此外,FDA 计划逐步取消对自制试剂项目的宽限,对其采用与其他IVDs相同的监管方案。由此可见,对于自制试剂项目的开展,尤其是高风险项目,需要更加严格的监管措施,以保证患者安全。目前,我国自制试剂仍处于起步阶段,相关监管措施还在探索中。
1.4 医疗机构建设多组学自制试剂实验室的必要性和可行性
自制试剂作为没有注册许可的IVDs 有着非常重要的临床意义。在美国,以下情况迫使一些医学中心或临床实验室通过自制试剂来应对临床紧急需求[4] :临床有紧急需求的检测项目, 但是市场上没有经过FDA 批准的CE-IVDs ;某些专业检测项目的市场份额太小,无法吸引商业参与;罕见病或孤儿病的检测由于相关试剂开发费用过于昂贵,只能依靠RUO ;经济效益有限的CE-IVDs 从市场上消失等。自制试剂主要集中在传染病检测、肿瘤学检测、人类基因检测、毒物和微量元素分析、罕见病或孤儿病检测等领域。
利用单一组学技术不能完全解读人类疾病的复杂性,基因组学、转录组学、蛋白质组学、代谢组学等多种组学数据相结合的多组学技术已成为一种应用日益广泛的用于理解遗传病生理病理机制的技术方法[8]。自制试剂覆盖范围很广,通常应用高通量测序技术、质谱技术、流式细胞分析等,对遗传性或获得性疾病相关基因型和表型、基因突变进行检测,用来评估高风险疾病和相对常见疾病[9]。在医疗机构建设多组学自制试剂实验室,不仅有助于弥补CE-IVDs 不足的困境,满足精准医学诊疗需求,还能通过多维组学技术组合对患者进行综合分析[10],从而更透彻地了解疾病病因,如分析更多的基因组,寻找和评估更佳的生物标记物和药物作用靶点,开发新的分子诊断工具,加快新疗法的应用,以及建立可靠的临床前实验模型,协助选择更为有利的精准治疗方式[11]。试点医疗机构具备完善的检验检测平台、专业的技术人员、完备的诊疗科目、高水平执业医生以及齐备的研究中心或科研职能部门,能够满足自制试剂从实验室检测到临床应用以及科研转化的全链条需求,推动多组学数据整合。
2、医疗机构多组学自制试剂实验室的建设实践
2.1 组织结构
2.1.1 成立专项工作组
多组学自制试剂实验室存在以下特点:使用自制试剂而非注册试剂,多个检测组在同一个实验平台工作,承担实验研究、临床检测和科研转化工作,需要医疗机构整盘管理和推动。中日友好医院成立自制试剂专项工作组,由主管院领导牵头,医务处、医学工程处、药学部、科技中心等相关职能部门参与其中。工作组整体统筹实验室建设筹备工作,在项目启动前进行评估和调研,提出人力、设备和场所需求;统筹人员和物资的调度,指导项目推进;制订实验室建设和运行方案,指导实验室的临床检测和科研工作。
2.1.2 建立自制试剂中心
自制试剂中心设立有检测部、质控部、科研与大数据部。检测部下设准入委员会和5 个检测组(感染性疾病检测组、个体化精准用药检测组、肿瘤液态活检检测组、质谱检测组、分子病理检测组),负责项目准入和临床检测工作。质控部下设质控专家组,负责监督和指导室内质控、室间质评和实验室间比对工作,同时负责项目退出。科研与大数据部下设生信团队和伦理委员会,负责生信分析、项目伦理审核和科研转化。
2.2 实验室和信息系统设计
2.2.1 实验室设计
依据《临床分子病理实验室二代基因测序检测专家共识》[12]相关要求,建设一个多组学的自制试剂实验室。实验室分区包括:试剂储存区、样本前处理区、试剂准备区、样本制备和提取区、文库制备区、杂交捕获区、多重PCR 区域( 第一扩增区)、文库扩增区、文库检测与质控区( 第二扩增区)、测序区、数据存储区。实验室不同组学实验平台依据检测样本和方法的差异进行有机整合,在前处理和建库时严格区分微生物样本、血液样本与组织样本;将测序实验室、基因芯片扫描室和质谱室分区设置;将数据存储和生信分析设在清洁区域。实验室建筑设计见图1。

2.2.2 信息系统设计
多组学自制试剂实验室作为新型实验室业务建设,其信息系统包括检测系统和生信系统。检测系统用于保证信息流的畅通,并且与院内现有系统高度集成,覆盖医嘱申请、条码打印、标本送检、标本签收、标本检测、测序平台对接、报告审核、结果查看和报告打印等环节。生信系统是基于测序数据库的数据分析平台,能够帮助自制试剂实验室高效存储、管理和分析大量测序数据,从而提高实验室的研究效率和科研成果产出速度。信息系统具体流程见图2。
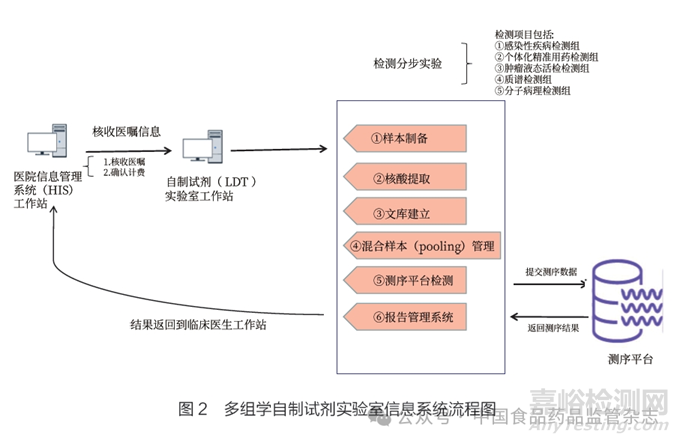
2.3 试剂制备和科研转化
自制试剂内部研发完成后,可以在医疗机构内部的生产车间进行制备,也可以委托具备条件的生产厂家进行制备。由于试剂制备车间有严格的分区管理和洁净度要求,医疗机构内部一般很难达到相关要求,通常以委托制备的方式进行。具体而言,医疗机构与有资质的生产厂家签订委托生产协议,并形成自制试剂委托生产管理制度,制定相关程序文件对委托制备厂家进行质量控制。试剂委托制备厂家资质审核和生产环节受药品监管部门的监督和管理。自制试剂中心的科研转化主要集中在将自制试剂转化为CE-IVDs,因此在项目准入时,自制试剂中心的科研与大数据部和医院的科技中心提前介入,协同完成学术和伦理审核,推进成果转化。
2.4 项目管理
2.4.1 项目准入
自制试剂中心的准入委员会负责新项目的准入评价,准入委员会由检验专家和临床专家组成。对于准入项目,由检测组提供项目的临床前确认文件,准入委员会依据项目的安全性、有效性、准确性和临床必要性等进行准入前评价[11],尤其要考虑项目的临床价值与临床可用性。获得实验室内部准入许可的项目通过学术委员会、伦理委员会和医疗委员会审批后,被呈报至上级药品监管部门备案审批。
2.4.2 项目质控
检测组严格按照样本处理、文库建立、上机测序、测序产物分析等相关流程开展检测工作,严格进行室内质控,以保障检测质量。自制试剂中心质控部指导室内质控、室间质评和实验室间比对。鉴于自制试剂的特殊性,大部分项目很难进行室间质评,可以通过比对不同实验室之间的相同项目积累项目质控经验。
2.4.3 项目退出
自制试剂项目开展的主要目的是解决紧急的临床问题,在临床应用过程中,可能出现安全性、准确性和有效性方面的问题。此时,由自制试剂中心质控专家组负责项目退出。出现以下情况之一的项目应退出:自制试剂被成功转化为CE-IVDs, 可以在不同实验室间推广使用;出现确定的质量问题等。此外,实验室间比对出现数据偏差的项目由质控专家进行分析。
3、对医疗机构多组学自制试剂实验室建设的建议与思考
3.1 实验室污染风险管控方案
多组学自制试剂实验室将微生物样本、血液样本和组织样本放在同一个实验室平台,进行第二代测序、病原微生物宏基因组测序、基因芯片扫描和质谱检测等复杂操作。在操作过程中,需要经过样本制备、提取、扩增、建库和测序等相关环节,容易出现样本间相互污染的情况。因此在实验室设计过程中,前处理和建库时微生物样本、血液样本与组织样本应严格分开;测序实验室、基因芯片扫描室和质谱室应分区设置;操作过程应严格按照单一动线走向;实验室内应安装紫外线灯、水浴箱等去污灭活设备。
3.2 项目风险管控方案
自制试剂为实验室自行研发、内部使用、自行制备或需要委托制备的试剂,在使用、知识产权保护等方面存在一定的风险。开展自制试剂项目的医疗机构需要制定严格的项目准入、自制试剂制备、实验室质控、项目退出和知识产权保护相关管理制度。自制试剂中心内部设准入委员会、质控专家组和伦理委员会等进行项目审核和风险排查;医疗机构通过学术委员会、医学工程处和科技中心指导自制试剂委托生产和科研转化。药品监管部门对相关项目进行备案审批,对自制试剂生产环节进行监管;卫生行政部门对实验室资质进行审核,并对检测环节进行监管。
3.3 信息风险管控方案
人类基因信息受法律保护,实验室测序信息只能控制在实验室内部。因此,在搭建实验室信息平台时,应当高度重视信息保护,只能使用院内系统。对于生信分析,在实验室建设前期,可能会使用其他公司的数据库,处理数据库信息时必须严格使用单机版系统。
自制试剂的产生是为了解决紧急医疗需求和注册试剂不匹配的矛盾,对于医学的进步和发展有非常重要的意义。自制试剂的发展和多组学技术的广泛应用,在为精准医学带来发展机遇的同时也带来了风险和挑战。我国自制试剂发展相对滞后,2021 年实施的新修订《医疗器械监督管理条例》允许符合条件的医疗机构开展自制试剂后,如何在发展自制试剂的同时有效防控风险是监管部门关注的重点。国家药品监督管理局会同国家卫生健康委员会将中日友好医院等10 家高水平医疗机构定为试点单位,以期提供“先行先试”经验。目前,中日友好医院已完成组织管理架构、实验室建设、信息系统建设、院内项目管理制度等前期试点工作。由于项目备案审批尚未完成,医院研发项目尚未投入临床运行,以上经验仅供其他医疗机构在自制试剂实验室建设初期借鉴。
引用本文
邬巧玲,赵建康,贾红兵,陈皇,高芃,张瑞苹,郗雅琪,杨瑞,阳玥,崔勇*.医疗机构多组学自制试剂实验室的建设实践与思考[J].中国食品药品监管,2024(3):28-33.

来源:中国食品药品监管杂志
关键词: 实验室自制试剂