
嘉峪检测网 2024-07-27 11:03
导读:本研究以采用无菌工艺生产的非最终灭菌冻干粉针剂为例,阐述无菌药品生产过程中潜在的污染风险,运用风险管理原则来制定有效的污染控制策略,以减少生产过程中的污染、保证无菌药品的质量。
无菌药品是指法定药品标准中列有无菌检查项目的制剂和原料药,包括无菌制剂和无菌原料药。无菌药品的生产须满足其质量和预定用途的要求,应当最大限度降低微生物、各种微粒和热原的污染,生产过程中防止污染是无菌药品生产的重大课题。本研究以采用无菌工艺生产的非最终灭菌冻干粉针剂为例,阐述无菌药品生产过程中潜在的污染风险,运用风险管理原则来制定有效的污染控制策略,以减少生产过程中的污染、保证无菌药品的质量。
无菌药品要求严格控制潜在的污染源,这些污染源可能以微粒、微生物或内毒素 (热原) 的形式存在[1]。《药品生产质量管理规范 (2010 年修订)》(GMP)《附录 1 无菌药品》中规定“无菌药品的生产必须满足其质量和预定用途的要求,应当最大限度降低微生物、各种微粒和热原的污染”[2]。因此在无菌药品生产过程中对各环节风险点制定有效的污染控制措施、优化风险管理方法,势在必行[3]。欧盟分别于 2017 年12月和2020年2月两次发布了《GMP 附录 1:无菌药品的生产》征求意见草案,该草案增加了污染控制策略 (contamination controlstrategy,CCS) 的要求 [4],并同时在 WHO 和国际药品认证合作组织 (PIC/S) 内联合征求意见。
我国目前尚无相关法规文件或指南要求在无菌药品生产中实施 CCS。欧盟《GMP 附录1:无菌药品的生产》指南中对 CCS 的定义是:源于对当前产品和工艺的理解,为确保工艺性能和产品质量,所计划的一套对微生物、热原和微粒的控制;这些控制包括与原料药、辅料、药品的物料和组分、设施和设备的操作条件、过程控制、成品质量标准相关的参数和属性,以及相关的监测和控制的方法及频次[5]。可见,CCS 是与无菌药品生产和质量相关的一整套对微粒、热原和微生物污染的控制措施和方法的总合,是基于产品和工艺知识、监管法规和生产企业质量体系的要求建立的,是要在整个产品生命周期中持续改进的。
冻干粉针剂作为无菌药品的常用剂型,采用非最终灭菌的无菌生产工艺,生产过程存在多因素的影响,相对于其他剂型属于高风险剂型[6]。因此,本研究利用 CCS 理念,采用风险管理原则对人流物流、环境、设施设备、生产工艺、无菌转移、卫生和消毒等生产各环节进行风险评估,期望有助于研究人员进一步识别并制定无菌药品生产过程的污染控制点。
1、冻干粉针剂生产过程质量控制点概述
冻干粉针剂通常的工艺流程见图1[7]。
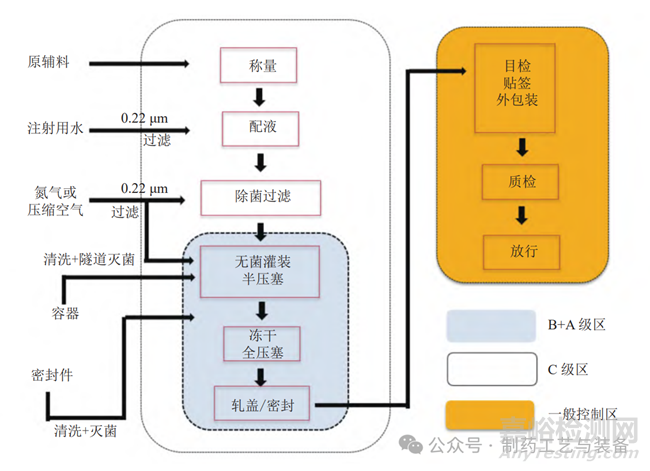
图1 冻干粉针剂生产工艺流程图
1.1 生产前清洗和准备 (含包装材料和工器具等)
冻干粉针剂生产用胶塞、西林瓶和工器具等的清洗灭菌场所均设置于 D 级或 C 级区域。生产企业应制定相应的标准操作程序 (SOP),明确规定包装材料的清洗和灭菌程序及相关参数,并对清洗和灭菌的效果进行验证和日常监控。
1.2 药液配制
药液配制是指按照相应品种的生产工艺规程,把原料、辅料等各种成分进行溶解配制,并按规定顺序混合制成待下一步灌装的药物溶液[8]。药液配制区域的洁净级别一般为 C 级。目前生产企业的配料系统已实现在线称量、pH 值检测、温度检测、在线清洗 (clean in place,CIP) 和在线灭菌 (sterilizein place,SIP)。药液配制过程要确保配制顺序正确,投料顺序、方式、参数等符合工艺规程。
1.3 除菌过滤
对于非最终灭菌工艺,除菌过滤是唯一的除菌手段[9]。通常采用二级过滤的方式过滤药液,使之达到 A/B 级;过滤器采用 SIP 进行灭菌,同时规定除菌滤芯在使用前后必须进行完整性测试等;还要对药液从配制结束到除菌过滤的间隔时间进行控制,时间跨度设定主要应从微生物控制以及药液稳定性两方面考虑。无菌保持时间通过培养基模拟灌装时间确定,药液稳定性通过产品工艺验证确认。
1.4 灌装、半压塞及自动进箱
无菌灌装区域处于 B 级背景下的 A 级区,该区域为非最终灭菌产品生产的高风险区域[10],须避免人员过多干预以及控制药品在该区域的暴露时间。灌装机通常要进行设备验证以提供稳定的性能,灌装机所在区域要设置限制进入屏障系统(restricted access barrier system,RABS),以有效保证 A 级区的洁净度、减少人员干预。经灌装、半压塞的产品,经过自动整列和进出冻干箱系统,转运至冻干箱内,整个过程均在 A 级区内自动完成,减少人员的干预。
1.5 冷冻干燥
冷冻干燥是指被干燥的含水物料在达到共晶点后凝固,然后在适当的真空度下升温升华,通过冷凝器将水蒸气冷凝,使物料低温脱水实现干燥[11]。冷冻干燥进出料区域处于 B 级背景下的 A 级区,冻干的过程包括冷冻、升华干燥和解析干燥 3 个阶段。目前冻干机通常能实现 CIP 和 SIP,冻干过程应确保各项参数符合产品生产工艺规程。冻干过程中所使用的气体一般通过 0.22 μm 过滤器除菌,其无菌性也是影响产品质量的关键性因素。
1.6 出箱轧盖
轧盖的目的是轧紧瓶颈处已全压的胶塞,从而保证长时间内包装系统的密封性和产品的无菌性。轧盖工序设置于 C 级或 B 级背景下的 A 级区,药品从冻干结束全压塞出箱到轧盖,全过程可采用自动化控制,杜绝人员干预对药品无菌性的影响。轧盖过程会产生大量的金属颗粒影响洁净区环境,所以特别要防止微粒污染。轧盖后要取样检查产品的密封完好性。通常,若轧盖设备带有自动剔废功能,则该工序可设在 C+A 级区,否则只能在 B+A 级区完成轧盖[12]。
1.7 目检、贴签和包装
完成轧盖的中间产品在普通区进行目检、贴签和外包装,目检方式一般有 3 种 :人工目检、半自动目检和自动目检。因产品在该工序阶段已处于密封状态,受污染的风险很小。
2、冻干粉针剂生产过程污染控制策略的涵盖内容
欧盟《GMP 附录 1:无菌药品的生产》指南 2.5章中提到,CCS 需考虑的要素包括但不限于:厂房和工艺的设计、厂房设施和设备、人员、公用系统、起始物料 (包括中间产品) 的控制、产品包装、供应商的批准 (例如关键组分的供应商、组件的灭菌及一次性使用系统和服务)、外包服务、工艺风险评估、工艺验证、预防性维护保养 (设备、公用系统及设施的计划性及非计划性维护保养)、清洁和消毒、监测系统和预防措施 (趋势分析、调查、纠正和预防措施)[5]。
CCS 的开发需要全面的技术和工艺知识[5],冻干粉针剂生产过程的污染来源包括环境污染来源[例如通过机械传播和空气传播带来的微粒、微生物和 (或)外来异物杂质污染]和交叉污染来源 (如通过混淆、残留、机械传播、空气传播等带来的物料和产品污染)。综合考虑欧盟 GMP 附录 1 指南中 CCS 的考虑要素和冻干粉针剂生产质量控制点,可从源头、时间限制、隔离保护和环境监控等四大方面控制其生产过程可能的污染来源,如表 1 所示。

表1 冻干粉针剂生产过程中控制污染的考虑要素
下面就冻干粉针剂生产过程中 CCS 的几个主要要素进行具体阐述分析。
2.1 源头控制
源头控制包括对物料、人员、设备、工具、容器的风险控制。
2.1.1 物料 (原料、辅料、包装材料等)
应对外购原辅料及包装材料制定供应商管理和现场审计制度,可对物料供应商实施分级管理,定期审计原料及内包材等关键物料,每年更新一次供应商资质材料;对物料的关键质控项目制定严于法定标准的验收标准或内控标准;制定物料的存储、发放和转移程序。
物料的内控标准应重点关注可能对产品质量产生影响的项目,包括细菌内毒素、微生物负荷、颗粒,其他项目有 pH 值、有关物质、含量等。
在物料存储管理中,应对已开口的物料进行评估和控制。针对风险较高的物料,如引湿性强、稳定性较差、使用频率较高的物料,应进行开口后存放时限的确认:模拟生产条件和存储条件,在开口 0、6、12 个月时取样检测,获取其质量变化情况,检测项目应包括污染控制的关键项目,如细菌内毒素、微生物负荷等,通过确认制定合理的存放时限。
直接接触药品的包装材料 (如胶塞、西林瓶等)通常存在 4 种污染:微生物、内毒素、微粒和化学污染 [13]。生产企业要按照相应的 SOP 进行清洗和灭菌,清洗和灭菌的方法应经过验证。通常,西林瓶采用干热灭菌 (320 ℃,5 min 以上),杀菌热力强度 (FH) 大于 1 365 ;胶塞采用湿热灭菌 (121 ℃,15 min 以上),F0 值大于 12。验证项目包括热分布、热穿透、细菌和 (或) 内毒素挑战等,确保除菌和除热原效果。同时,与药品直接接触的包装材料和容器具的清洗、干燥和灭菌的间隔时间应该尽可能短,并应严格控制灭菌后至使用的存放时限,对于灭菌后的物品需要进行密闭保存,避免清洗和灭菌后的物品被二次污染。
2.1.2 水系统
水是冻干粉针剂生产的主要溶媒、清洁介质。水系统要定期进行化学和微生物监测,并基于对持续监测数据的确认和回顾制定警戒限度,以识别系统性的不良趋势 [14]。取样计划应反映 CCS 的要求,包括:①所有用水点在规定的时间间隔内全部被取样,确保定期取得具有代表性的水样进行分析;②潜在的最差取样位置,如最远端的用水点、回水点等;③每天配液用水点的取样。
2.1.3 人员
人是最大的污染源,因此必须针对无菌操作人员建立有效的污染控制策略 [15]。
①规定关键区域允许进入的操作人员的最大数量,通常 B 级区不超过 8 人。制定不同洁净区人员的更衣要求,并且人员必须经更衣确认后方可获得授权进入洁净区。洁净区门口应建立人脸识别、指纹识别、凭密码或门禁卡进入的设施,限制人员随意出入。外来人员应在经批准后、由管理人员陪同进入洁净区。
②制定允许或取消人员进入洁净区资质的体系,制定洁净区行为规范,并对洁净区工作人员进行微生物知识、洁净区无菌及微粒控制、洁净区行为规范等内容的培训,并做好记录供查询。
③ B 级洁净区人员的着装要求:全身密闭,佩戴眼罩、手套等 (见图2),每班生产结束后采集表面微生物进行监测。

图2 B级洁净区人员着装图
2.1.4 工器具及容器
建立工器具和容器清洁 SOP,规定清洁程序的相关参数,如水温、流速、清洁时间,清洁后目视检查应无肉眼可见残留,并对清洁程序进行验证 [16]。
容器具设置标识管理,状态标识信息一般包含以下项目:清洁 / 灭菌、有效期、操作人等;并按色标管理,一般情况下,已清洁灭菌采用绿色标识、已清洁未灭菌采用黄色标识、未清洁灭菌采用红色标识。
针对进入 B 级区的容器具要建立相应 SOP,明确规定需要进行灭菌的容器具的装载方式,且该装载方式应经过确认并以图片形式贴在设备旁边,以便操作人员按照既定的装载方式进行灭菌。规定的灭菌程序应经过验证;灭菌过程应记录关键参数,并采用自动打印方式保存。
2.2 时间限制
微生物无处不在,而且是动态生长的,以几何级数裂变繁殖,因此时间限制是控制微生物数量所必要的 [17-18]。无菌药品生产需要规定设备使用、物料使用及工序操作的时限,生产过程各个环节的持续时间应限定在规定的最大时间限度内,该时间限度应经过验证,包括:①设备、组件和容器的待清洁、清洁干燥后存放的最长时限;②灭菌后设备、组件和容器在使用前和组装或灌装期间能保存的最长时限;③洁净环境如 RABS 和隔离器 (isolator),在组装或灌装期间的最长保持时限;④产品配制开始到除菌过滤的最长时限;⑤除菌过滤后到最终无菌灌装工序结束之间的最长时限;⑥灭菌后容器和密封组件在关键操作区域 (包括灌装区) 内脱包前的最大保留时限以及脱包后的最大暴露时间;⑦胶塞清洗干燥至灭菌的最长放置时间,以及灭菌后胶塞的最长放置时间;⑧无菌操作的最长时限。
2.2.1 设备保持时限
生产设备使用后需要考虑 2 个保持时间 :生产结束后到设备开始清洗的时间段 (污染保持时间,dirty hold time) 和设备清洗后到设备再次使用的时间段 (清洁保持时间,clean hold time),两者都需要进行确认。通常污染保持时间主要考虑活性物质和微生物在放置一段时间后按既定的清洁程序能否被有效清洁,其验证项目包括活性成分的残留、微生物负荷、总有机碳和电导率等。
污染保持时间应该在基本的清洁验证方案中给予评价,并作为一个最差条件或挑战;清洁保持时间可作为基本清洁验证方案的第二部分,或作为一个单独的方案与基本清洁验证方案分开。清洁验证研究和清洁保持时间研究具有一定关联性;例如,清洁结束时的微生物负载数据也可作为清洁保持研究的起始微生物负载数据。
2.2.2 物料时限
物料时限的项目包括生产用原辅料、生产过程的中间产品、生产用清洁剂和消毒剂的保存时限等。
要确定生产用原辅料的保存时限,如应对温度敏感固体物料的转运和暂存时限,以及生产用液体物料的配制和存放时限进行考察和验证。
建立温度敏感固体物料的转运、暂存、传递和使用 SOP,日常监控温度,并经过适当验证。对于生产用液体物料,应制定转运、暂存、配制和过滤SOP,日常监控并经过验证;重点验证过滤时间,包括除菌过滤前保留时间、除菌过滤时间、除菌过滤后存放时间。
生产用清洁剂、消毒剂的保存时限,包括未开封清洁剂、消毒剂的保存期限,配制后清洁剂、消毒剂的保存期限,以及开始使用后清洁剂、消毒剂的保存时限,同时确认保存期限最长的消毒剂的消毒效果。例如:对于开封后消毒剂存放时限的确认,可模拟生产使用状态,在开启后 0、12、24、48、72 h 时分别取样测定其抑菌效能,确认消毒剂在生产状态下最长的保存时限。
2.2.3 工序操作时限
工序操作时限可从以下方面考虑,工序操作的最长时限、物料或产品暴露的最长时限、设备或环境使用的最长时限。
在起草工艺验证或清洁验证方案时,应考虑在最差的工艺条件下完成 3 次验证评价。最差工艺条件的原则包含或引用在验证方案中;最差工艺条件,包括最长的生产保持时间、阶段性生产中最大批量或最长的运行时间、最短的清洗时间、最差清洗的温度和最差的 CIP 循环程序等。例如:①灭菌后的设备、组件和容器在使用前和组装或灌装期间的保存时间;②洁净环境如 RABS 和隔离器,在灌装或组装期间的保持时间;③制定产品从配制开始和经过除菌过滤到最终无菌灌装工序结束之间的最长时限,包括无菌组装、无菌操作时间、灌装时间;灭菌后容器和密封组件在关键操作区域 (包括灌装)内密封前的最大暴露时间;胶塞清洗干燥至灭菌的放置时间、灭菌后胶塞和密封件的放置时间。
2.3 隔离保护[19]
生产过程采用 RABS 和隔离器 2 种方式,确保无菌所需的条件,最大程度减少无菌关键区域中与人为接触干预相关的微生物污染。
2.3.1 限制进入隔离系统 (RABS)
RABS 可提供封闭但未密封的环境。人员操作时,使用手套和其他集成的传输端口执行操作或将材料输入内部;根据设计的不同,门很少或不能打开。RABS 包括开放式限制进入隔离系统 (openrestricted access barrier system,ORABS) 和密闭式限制进入隔离系统 (closed restricted access barriersystem,CRABS),见图 3。

图3 ORABS(A) 和 CRABS(B) 的外观
ORABS 采用独立的高效空气过滤器 (highefficiency particulate air filter,HEPA) 和供热通风与空气调节 (heating ventilation and air conditioning,HVAC) 系统,舱内空气排入 B 级洁净室内 (见图4A),采风部分或全部来自室内 B 级区,舱内温湿度及单向层流受到室内空气影响,空气不可以在ORABS 内循环,且不可以生产毒性大、活性高的产品,对操作者的防护效果低。
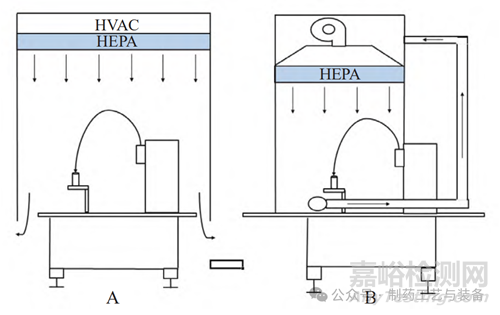
图4 ORABS(A) 和 CRABS(B) 的空气流型示意图
CRABS 是介于 ORABS 和隔离器的过渡设计,舱内空气可经处理后循环使用 (见图 4B),不排入B 级背景区,可降低 HVAC 系统的能耗,能生产毒性大、活性高的产品,对操作者的防护效果高。
2.3.2 隔离器 (isolator)
隔离器提供了内部工作区域,是能满足 A 级区空气质量的净化装置,可以为其内部提供与外部环境无间断的持续隔离。系统通过与辅助设备无菌连接来实现物料的转移,而不是使用向周围环境开口的方式,因而能避免隔离器内部被外部污染,使整个系统在操作过程中始终保持密闭状态。
隔离器可在称量、投料、转移、灌装、冻干、轧盖等生产环节中应用。
2.4 环境监控
环境及人员的活动会产生大量的微粒,并悬浮于空气中;而冻干粉针剂的配制、灌装是完全暴露在空气中的,所以环境空气质量会直接影响无菌药品的澄明度。因此,需加强环境空气中非生物微粒的监控[20]。
①建立持续可靠的环境监测程序,例如确定合适的取样位置、监测频率、监控方法和培养条件 [如温度、时间、有氧和 (或) 无氧条件];通过监测过程中获得的监控数据和所发现的微生物菌群,进行无菌环境的综合评估并找出风险点;加强日常的环境监控,同时采取必要的预防和纠正措施,确保无菌环境和生产产品的质量。通常环境监控包括 A级环境和 B 级背景环境的沉降菌、浮游菌、设备表面菌等,监控频率可每班一次,包含从无菌组装开始到生产结束的整个过程。
②制定良好的监测计划,结合空调系统的清洗、清洁和消毒周期,制定监控的合适位置和监测频率。选择的取样监测点和取样频率应能够鉴别出系统潜在的风险,并能根据情况 (如生产操作改变、法规变更、微生物趋势变化、增加新设备、公用系统改造等),做出临时或长久调整。
③为确保 A 级关键区域能够提供合格的气流用于保护产品生产,需要定期对 A 级区域进行空气流型的确认,例如进行烟雾试验,以确保所有无菌开口操作的位置都能够得到有效的气流保护,避免产品污染。
另外,除上述 CCS 外,生产过程还需设置不同的环境级别,如外围、生产区、受控不分级(controlled not classified ,CNC) 区、D 级、C 级、B 级及 A 级,使人员、物料及工器具容器逐级由低级别环境进入高级别环境,在整个转移过程 (包括清洁控制、脱外包、消毒净化、灭菌等) 中,污染梯度递减,直至满足无菌的要求 [21]。
3、冻干粉针剂的污染控制策略总结
为确保无菌冻干生产工艺的性能和产品质量,一个完整 CCS 的内容框架通常包括以下几个方面:车间和产品的基本信息、污染控制的风险评估和控制、污染控制措施的效果评价。
3.1 车间和产品的基本信息
车间的基本情况:无菌冻干粉针剂车间布局,洁净级别设计及洁净区分布、人流物流流向、压差和气流流向。
生产设备的基本情况:关键的生产设备清单,称量及配液设备、洗瓶机、灌装机、冻干机、轧盖机、灯检机等关键设备的信息,层流罩、隔离器、空调净化系统等信息。
产品的基本情况:无菌冻干产品的工艺路线、关键工艺控制点、工艺中污染的关键控制点、除菌过滤前的生物负载、除菌过滤器的完整性测试、灭菌包装材料的传递、分装过程的环境条件控制、包装过程的密封性保障等。
人员的基本情况:生产操作分为几个班组,每班的最长工作时间,人员操作的日常监控情况、无菌更衣确认。
3.2 污染控制的风险评估和控制
基于对硬件和软件的了解和对工艺过程的认知,对潜在污染的风险进行识别和评估以及采取控制措施,主要包括:无菌冻干的工艺流程和车间设计、设施和设备安装、人员资质和培训、公用系统设计和运行、产品包装和密封、清洁消毒、环境监控和趋势分析等。
3.3 污染控制措施的效果评价
当实施了污染控制策略以后,应根据得到的数据或结果,评估对应的风险是否得到了有效控制。若效果评价认可,则定期再评价;若效果评价不认可,则要重新评估并更新控制策略,再评价。相关部分发生变更时,也应更新评估、控制策略和效果评价的相应内容。
参考文献
[1] 曾冬娇.新版GMP中对无菌工艺验证要求的探讨[J].中国战略新兴产业: 理论版, 2019, (21): 1.
[2] 国家药品监督管理局.《药品生产质量管理规范(2010年修订)》附录1:无菌药品[EB/OL].[2020-02-23].https://www.nmpa.gov.cn/directory/web/nmpa/images/MS7O3r760qnGty5kb2M=.doc.
[3] 郑明泽.关于GMP在无菌药品生产质量管理中细节问题的应用与分析[J].临床医药文献电子杂志, 2019, 6(30):195.
[4] Anon.Second targeted stakeholders' consultation on therevision of Annex 1, on manufacturing of sterile medicinalproducts, of Eudralex volume 4 [EB/OL].[2020-02-23].https://ec.europa.eu/health/medicinal_products/consultations/2020_sterile_medicinal_products_en.
[5] Anon.Annex 1: manufacture of sterile products [EB/OL].[2020-02-23].https://ec.europa.eu/health/sites/health/files/files/gmp/2020_annex1ps_sterile_medicinal_products_en.pdf.
[6] 米海生.冻干粉针剂在生产过程中的质量控制点解析[J].中国化工贸易, 2019, 11(1): 235.
[7] 陈王露, 詹星星, 孙志勇.浅议冻干粉针剂生产中可见异物的来源及其控制措施[J].机电信息, 2018, (11): 46-48.
[8] 李邢珺.冻干粉针剂生产过程的质量控制[J].缔客世界,2019, (7): 21.
[9] 李 娜, 韩建军.无菌药品除菌用滤芯的考察[J].文渊:中学版, 2018, (11): 378.
[10] 王 健.无菌工艺模拟灌装方法的优化[J].东西南北: 教育, 2017, (9): 12.
[11] 武丹丹.真空冷冻干燥技术在生物制药方面的应用分析[J].中国科技期刊数据库 科研, 2017, (2): 33.
[12] 泰凌生物制药江苏有限公司.一种带废屑收集的轧盖机:中国, 207404806U [P].2018-05-25.
[13] 王 俊.小容量注射液生产工艺管理要点探讨[J].临床医药文献电子杂志, 2017, 4(84): 16624.
[14] 徐昕玥, 梁 毅.制药用水系统警戒限度与纠偏限度的制定与管理[J].机电信息, 2017, (17): 6-8.
[15] 李建国.探析无菌药品生产过程中的无菌控制[J].家庭医药, 2019, (6): 216-217.
[16] 芦旭升.药品生产设备清洁验证关键点的研究[J].科学与财富, 2017, (25): 262.
[17] 丁 芬.注射剂微生物污染水平测试及工艺时限确定[J].中国科技投资, 2018, (23): 252.
[18] 陈传就.浅谈注射剂生产的微生物控制[J].医学信息,2016, 29(8): 348-349.
[19] 张 伟.制药行业的无菌隔离技术探讨[J].中国科技投资, 2019, (30): 243.
[20] 陈春凌.新版GMP无菌药品生产环境监测探索[J].世界最新医学信息文摘, 2017, (41): 169.
[21] 张银花.FMEA法在冻干粉针剂生产过程中的风险管控[D].延吉: 延边大学硕士学位论文, 2017.
本文作者李国琼、翁贤坤、梁玉琴,海南省药品审核认证管理中心、海南倍特药业有限公司,来源于中国医药工业杂志,仅供交流学习。

来源:Internet
关键词: 冻干粉针剂