
嘉峪检测网 2024-11-20 19:58
导读:本文介绍了GMP文件管理体系在生产车间实际运用。
新版GMP(2010版) ,对医药行业生产管理中文件管理部分做了详细的描述与介绍,生产车间以此为依据,编写岗位操作规程等,并在实践中得到运用,对车间实际运用的积极作用。
文件管理是质量管理系统的基本组成部分,能够使企业在从事任何活动时,都能够做到并遵循有法可依,可以最根本的满足日常生产、日常检验、上市销售等生物链的质量管理手段合规划的手段。通过质量系统文件的实施来保证质量体系的有效运行。
中国 GMP(2010版) 第八章对文件管理的要求进行了描述。与中国 GMP(1998版) 相比较,增加了质量标准、批包装记录的具体要求; 对文件管理的基本要求、工艺规程和批生产记录的内容进行了更为详尽的描述。
1、新版GMP文件管理体系结构
生产企业的文件体系均可以按照生产管理,质量管理情况进行分类,也可以按照 SOP 文件系统的架构对文件体系进行分类,可以分为文件管理类文件、质量体系类文件、操作规程类文件和书写记录类。(表1) 。
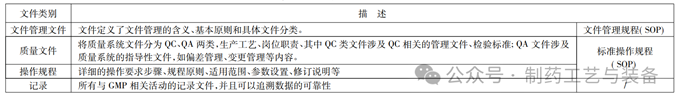
表1 文件管理体系分类
根据公司的生产体系、质量体系、产品特性的不同,可以将公司的文件按照类别进行分,可以有公司的组织架构、岗位职责、质量标准、生产类的岗位操作规程等,也可以将质量体系文件分为两类,一类是 QC 文件,另一类是 QA 文件; 其中 QC 文件可以将检验方法、质量标准分开,QA 文件可以将所有质量保证类的如偏差,变更等均统一放在一类。
2、文件管理的生命周期
生产车间所有使用的文件,如工艺规程、操作规程(物料类、卫生类) 、设备类等所有的文件均有生命周期。生产车间使用的工艺规程、岗位操作规程等文件,均需要满足在整个生命周期的合规性和满足生产需求。
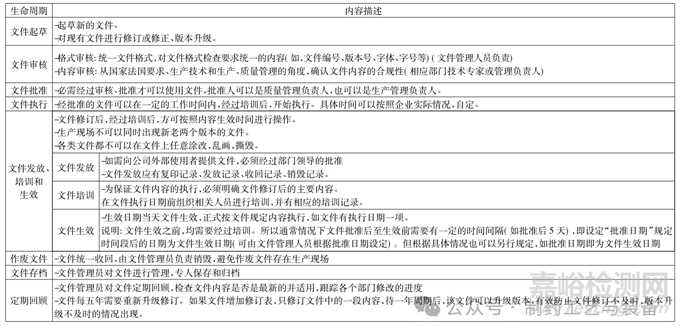
表2 生命周期的介绍分析
在使用过程中,可以通过对生产车间的产品进行工艺验证、验证产品的清洁验证、新增设备验证等,不断的完善 SOP 内容,细化步骤,健全内容,使文件在整个生命周期内,始终是最新的,操作性最强的,最符合法规要求的 SOP。
生产车间可以根据自身实际情况,在车间内部设置专人管理文件(文件管理员) ,负责对文件进行起草、修订、排版等工作; 可以有效满足生产过程中,出现修订文件立即修订的情况。
3、文件保存期限
(1) 批相关的文件。批生产记录和批包装记录均由质量管理部门专人负责管理统一保存,至少保存至药品有效期后一年,并且要有销毁记录。
(2) 其他类别的相关的文件。依据 GMP 规定要求,产品的质量标准、工艺规程、操作规程、稳定性考察、确认、验证、变更等其他重要文件应当长期保存,并且需要保存原件,不得销毁。对于其他文件记录 GMP 无明确规定的,生产企业的车间可以按照麻、精、特药等实际生产、产品销售链条的情况特点等因素,所有与药品相关的生产记录、台账、票据均需要制定相应的保存年限,保证产品生产、质量控制和质量保证等活动可以追溯。
4、文件种类
(1) 药品的质量标准。药品的质量标准对于生产车间来说,在中间体和半成品的过程控制起到关键性意义,但是质量标准的修订,对国家政策法规的把握与运用,则由质量管理部负责。中国 GMP(2010 版) 中要求: 物料和成品应当有经批准的现行质量标准; 必要时,中间产品或待包装产品也应当有质量标准。按照 GMP 条款内容要求,企业生产的所有产品的生产都必须有检验使用的质量标准、国标、部标或注册标准。企业质量部门在起草质量标准时,必须遵循国标、部标或注册标准,企业可以在中间产品环节设定内控标准,遵循就高不就低的原则,最终目的也是严把质量关。
(2) 工艺规程。中国 GMP(2010 版) 中要求: 工艺规程的制定应当以注册批准工艺为依据。工艺规程不得任意更改。所有产品的工艺规程起草,必须遵循注册生产工艺的内容,不得擅自变更,如若变更生产工艺,需要向国家药监局提交申请等证明性资料,生产车间不可以随意更改生产操作步骤,生产关键操作参数等内容。
(3) 批记录。中国 GMP(2010 版) 中要求: 批记录用于记述每批药品生产、质量检验和放行审核的所有文件和记录,可追溯所有与成品质量有关的历史信息。生产车间的批记录包括: 批生产记录、批包装记录。质量管理部的产品批检验记录、审核结束后,由填写放行审核记录并将复核,该批次批生产记录、批包装记录、批检验记录有无发生偏差,若有偏差发生,必须彻底调查清楚后,方可对本批产品给予放行。批生产记录、批包装记录和批检验记录的起草必须严格按照已注册批准的生产产品生产工艺进行起草,不得擅自更改工艺流程、工艺参数、工艺内容的。
5、记 录
生产车间使用的记录主要包括生产相关房间的温湿度记录、压差记录、岗位的设备点检表、设备使用日志、设备维护保养记录、模具管理记录等辅助记录。任何书写的记录都是反应实际生产活动实施结果的书面文件,药品生产的所有环节,从生产到检验到销售都要记录可查证追溯。
6、任何部门和生产车间在编写文件的时候,需要注意
任何部门和车间,在起草文件、修订文件的时候,必须遵循一切内容的真实性,法规的符合性,必须遵循产品的工艺要求、生产环境要求。文件的起草者,可以是每个使用部门指定的专人起草,并有该部门负责人对文件进行审核,当审核通过后,可由质量负责人或生产企业负责人批准生效。所有的文件原版文件可以由质量部门负责文件的保管、复制、分发等管理。
(1) 质量标准的制定依据。生产企业使用的原料、辅料质量标准的制定可以参照《中国药典》、包装材料质量标准的制定可以参考《24 号令》,中间体(半成品) 、成品均也有质量标准。有国际法定标准的要按照国家法定标准进行编写 (如《药典》) ,无国家法定标准的按国家行业标准、地方标准等进行编写。中间体( 半成品) 、成品需要制定企业内控标准,企业内控标准不得低于法定标准。物料和成品质量标准编写时,应包括的内容见下表,表3。
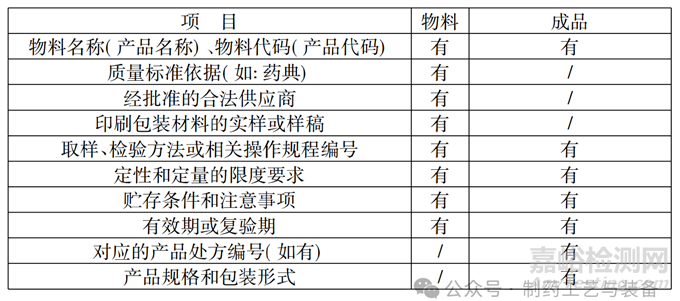
表3 物料和成品质量标准应包含内容
从上表中可以明确的看到,物料代码(产品代码) 已经作为企业管理的一项要求。物料代码(产品代码) 具有专一性,从物料采购进仓库后,就根据企业的编订的编码原则,对物料进行编码,所有物料均需要编码。每一个产品,根据不同的剂型,编订产品代码,涉及产品代码的文件、记录都需要详细记录,保证产品使用文件中涉及的产品代码与记录保证统一性、正确无误,以便于产品的追溯。
(2) 起草、修订工艺规程的要求。新产品在起草工艺规程时,所有的产品工艺步骤,操作控制要求要点均处于摸索阶段,通过三批工艺验证,最终确定工艺步骤、操作关键点、参数设置范围等,而此时产品的工艺规程在工艺验证前,可以称之为草案,待工艺最终确定后,可以升级文件版本,称其为某产品的工艺规程。
(3) 批记录的分类。生产车间使用的批记录可以分开为批生产记录、批包装记录、批检验记录,可以将批生产记录与批包装记录合二为一,也可以单独分开。批生产记录和批包装记录,车间可以由专人负责修订,如车间技术员。批检验记录则可以由质量管理部 QC 专人负责。
(4) 操作规程和记录。生产车间涉及操作规程的应当均有详细的记录,质量标准类的操作规程、生产过程的操作规程、工艺规程、批生产记录管理规程、批包装记录管理规程、文件管理类的操作规程,均可以设计出文件表头,需要包括文件名称、起草者、审核者、批准者、起草日期、审核日期、批准日期、执行日期,文件编号、版本号、分发部门等,公司文件统一格式,并加注公司 Logo,可以使文件视觉上更美观。企业结合实际生产管理,产品特性,生产销售量的实际情况,需要依据中国 GMP(2010 版) 要求,进行操作规程的编写,以及日常工作的详细记录,按照生产计划,科学、合理、有效的完成操作规程,及时记录数据,并进行相关的风险评估管理。
7、结 语
所有文件的起草,均依据新版 GMP 来制定。文件的内容应清晰、易懂,并有助于追溯每批产品的历史情况,所有的生产活动均需要通过文件,记录来体现证明。文件是保证产品生产过程的指南针,是质量保证的根本,只有科学的使用文件,制定文件,才能科学有效的保证产品质量,才能使患者使用到放心的产品。
参考文献:
[1]《药品生产质量管理规范及附录》(2010 年修订) .
[2]《药品 GMP 指南》.中国医药科技出版社,2011,8(第一版) .
本文作者周冉,江苏恩华药业股份有限公司,来源于科技风,仅供交流学习。

来源:Internet
关键词: GMP文件