
嘉峪检测网 2024-12-03 08:42
导读:尽管GMP的基本要求是只有在最终检测完成后才放行批次,并且在药品指南中有明确的描述,但FDA检查员在现场检查时,在某些情况下,在成品质量控制方面一再遇到严重的GMP违规行为。
In spite of the fact that it is a basic GMP requirement to release a batch only after final testing has been carried out, and although this is described clearly and explicitly in the pharmaceutical guidelines, FDA inspectors repeatedly encounter in some cases serious GMP violations in the area of quality control of finished products when they carry out inspections on site.
尽管GMP的基本要求是只有在最终检测完成后才放行批次,并且在药品指南中有明确的描述,但FDA检查员在现场检查时,在某些情况下,在成品质量控制方面一再遇到严重的GMP违规行为。
The following chart shows the percentage of Warning Letters addressed to manufacturers of drug products because of deficiencies concerning the requirements of 211.165 over a period of five years.
下表显示了在五年内,由于放行检测(21 CFR 211.165,法规内容见文末)的缺陷而致药品生产商的警告信的百分比。
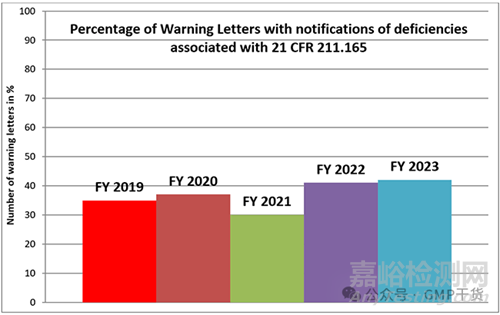
Figure 1: Percentage of Warning Letters with notifications of deficiencies associated with the testing of the finished product according to 21 CFR 211.165 for the fiscal years 2019 - 2023
图1:根据2019-2023财政年度21 CFR 211.165,含有与成品测试相关缺陷通知的警告信百分比
The percentage of Warning Letters citing 211.165 has settled at a little over 40% in the last two fiscal years. In absolute numbers this means 30 Warning Letters in the FY 2023 and 18 in the FY 2022.
在过去两个财政年度,引用211.165条款的警告信比例稳定在略高于40%的水平。在绝对数字上,在2023财年有30封警告信,2022财年有18封警告信。
Categorisation of GMP Deficiencies Associated with the Release Testing of the Finished Product
与成品放行检测相关的GMP缺陷分类
The deficiencies described in the Warning Letters can be divided into the following categories:
警告信中所述的缺陷可分为以下几类:
No Microbiological Testing
没有微生物测试
One of the GMP deficiencies cited most often in the Waning Letters is the lack of testing of the finished product for objectionable microorganisms according to 211.165(b). FDA inspectors have observed this especially at producers of hand sanitizers.
在警告信中最常引用的GMP缺陷之一是未根据211.165(b)对成品进行不良微生物检测。FDA检查员发现,这种情况在洗手液生产商身上尤为明显。
Incomplete Release Testing
不完整的放行检测
This deficiency is described nearly as often. Contrary to the requirements of 211.165(a) according to which release testing shall include testing for compliance with the specifications of the finished product including identity and strength of the active ingredient, manufacturers have released their drug products on the basis of incomplete testing (such as odour, appearance or other physical parameters). In single cases the product was even released prior to the receipt of the test results.
这种缺陷几乎经常被描述。与211.165(a)的要求相反,放行检测应包括符合成品规格的测试,包括活性成分的特性和强度,生产商在不完整的测试结果(如气味、外观或其他物理参数)的基础上放行了他们的药品。在个别情况下,产品甚至在收到测试结果之前就被放行了。
The lack of testing of the batches of the finished product for impurities is also described several times in the Warning Letters.
在警告信中也多次描述了成品批次缺乏杂质检测的情况。
No Acceptance of Responsibility for the Release Testing
不承担放行检测的责任
Several contract companies finishing the product for instance by only filling it up have transferred the responsibility for release testing to the customer. But according to the FDA the production site carrying out the last manufacturing step immediately prior to the release of the drug product is obliged to carry out the batch release. FDA considers the transferral of responsibility to be a GMP violation.
一些CDMO生产了产品,例如只进行灌装,然后把放行检测的责任转移给了客户。但根据FDA的规定,在药品放行前进行最后一个生产步骤的生产现场有义务进行批放行。FDA认为责任转移是违反GMP的。
No Rejection of the Batch in Case of a Failed Release Testing
在放行检测失败的情况下,未拒绝该批产品
In several cases the quality control unit did not reject a batch or place a hold on it after the final testing failed.
在一些情况下,质量部门在最终检测失败后没有拒绝批次或将其冻结。
Use of Non-validated Methods for the Release Testing
使用未经验证的方法进行放行检测
According to 211.165(e) only validated methods may be used for release testing. In some cases, FDA inspectors discovered that the quality control units released batches of the finished product after testing with non-validated methods.
根据211.165(e),只有经过验证的方法才能用于放行检测。在某些情况下,FDA检查员发现质量部门在使用未经验证的方法进行测试后放行了成品批次。
Follow-up Requests from the FDA
FDA的后续要求
Usually Warning Letters contain a follow-up request for documents/evidence in respect to each GMP deficiency associated with one of the paragraphs of CFR 211. In the case of 211.165 the FDA demands the following:
通常警告信包含对与CFR 211段相关的每个GMP缺陷的文件/证据的后续要求。在211.165案例中,FDA要求如下:
A list of chemical and microbial specificationsof the finished product, including the relevant test methods.
成品的化学和微生物标准清单,包括相关的测试方法。
Anaction plan and timelines for conducting full chemical and microbiological testing of retain samples of all batches distributed to the United States that are within expiry as of the date of the Warning Letter.
一份行动计划和时间表,用于对所有在警告信有效期内分销到美国的批次的留样进行全面的化学和微生物测试。
Asummary of the results obtained from testing retain samples.
留样检测结果摘要。
Acomprehensive independent assessment of the methods, procedures, equipment, documentation of the quality control unit, and analyst competencies. Based on this review, provision of a detailed plan to remediate the deficiencies and evaluation of the effectiveness of the laboratory system.
对质量控制部门的方法、程序、设备、文件和分析人员能力进行全面的独立评估。在此审查的基础上,提供详细的计划,以纠正缺陷和评估实验室系统的有效性。
If these proofs can be provided in the course of an FDA inspection the topic „release testing“ shouldn‘t pose a problem.
如果这些证据可以在FDA检查过程中提供,那么“放行检测”应该不会构成问题。
与放行检测相关的法规要求
FDA对成品批次放行测试的要求如联邦法规21 CFR 211.165所述,测试和放行分销是强制性的。该条款的六节载有下列规定:
(a)每批成品放行前应进行符合规格要求的测试。这包括测试活性成分的特性和强度。如果对寿命较短的放射性药物进行无菌和/或热原测试,只要尽快进行测试,这些放射性药物可以在测试结果出来之前放行。
(b) 每批药品应进行适当的实验室检测,以确保无不良微生物。
(c) 任何取样和测试计划应以书面程序描述,其中应包括取样方法和每批待测试的样品数量。应遵循上述书面程序。
(d) 质量部门进行的取样和检测的接受标准应足以确保药品批次符合每一适当的规格和适当的统计质量控制标准,这是放行他们的条件。统计质量控制标准应包括适当的接受水平和/或适当的拒绝水平。
(e) 所采用的检测方法的准确性、灵敏度、特异性和可重复性应建立并形成文件。此类验证和文件可以按照§211.194(a)(2)完成。
(f) 不符合制定的标准、规范和其他有关质量控制标准的药品应当予以拒收。可以进行再加工。在接受和使用之前,再加工的产品必须符合适当的质量标准、规格和任何其他相关标准。
以下检查指南包含了FDA检查员在现场工作的指导方针:
FDA药品质量控制实验室检查指南(1993年7月)
FDA微生物药品质量控制实验室检查指南(1993年7月

来源:GMP干货
关键词: 放行检测