
嘉峪检测网 2025-06-09 19:14
导读:本文针对口服固体制剂中的不溶性杂质,建立了:高效液相色谱(High-PerformanceLiquid Chromatography,HPLC)、扫描电子显微镜(Scanning Electron Microscope,SEM)和激光粒度分析相结合的方法,检测不同批次制剂中不溶性杂质的含量、颗粒分布及其对药物释放度和稳定性的影响,为优化制剂质量控制体系和提升药品标准化生产提供科学依据。
目的 研究优化口服固体制剂中不溶性杂质的检测方法,并评估杂质对制剂稳定性的影响。
方法 采用高效液相色谱(High-PerformanceLiquidChromatography,HPLC)、扫描电子显微镜(Scanning Electron Microscope,SEM)和激光粒度分析技术,测定不同批次口服固体制剂中不溶性杂质的含量、颗粒分布及药物释放度。通过加速稳定性试验,考察样品在室温(25℃)和高温高湿(40℃,75%RH)条件下存储6个月后的杂质变化、药物释放度及外观稳定性,评估不溶性杂质对制剂质量的影响。
结果 实验结果显示,各批次样品中的不溶性杂质含量符合药典标准,颗粒粒径分布均匀,且不溶性杂质对药物释放度影响较小。通过6个月的稳定性测试,制剂在存储过程中未出现显著变化。结论 通过优化的检测方法,可以有效评估不溶性杂质的含量与分布情况以及制剂的质量稳定性,为口服固体制剂的质量控制提供了科学依据。
口服固体制剂广泛应用于临床治疗中,其质量直接关系到药物的疗效与患者的用药安全。制剂中的不溶性杂质会影响药物的稳定性和释放度,因此,研究不溶性杂质的检测方法及其对制剂稳定性的影响,具有重要的理论和实践意义。本文针对口服固体制剂中的不溶性杂质,建立了:高效液相色谱(High-PerformanceLiquid Chromatography,HPLC)、扫描电子显微镜(Scanning Electron Microscope,SEM)和激光粒度分析相结合的方法,检测不同批次制剂中不溶性杂质的含量、颗粒分布及其对药物释放度和稳定性的影响,为优化制剂质量控制体系和提升药品标准化生产提供科学依据。
Part.01材料与方法
1.1 实验材料
本研究所用的实验材料包括口服固体制剂样品、试剂及标准品。口服固体制剂样品选取了市售的多个批次药品。所用原料药均为国内某知名制药企业生产的合格产品,且符合药典规定的质量标准。所有试剂均采用分析纯级别,液相色谱使用的溶剂均为高纯度乙腈、甲醇和去离子水,确保无干扰杂质。标准品的选择依据药典或国际药品标准,确保了实验测定的可靠性。为了提高数据的代表性,选取的样品覆盖了不同生产批次。
1.2 实验设备与工具
本研究使用了Agilent 1260 Infinity II高效液相色谱系统(美国安捷伦公司)进行不溶性杂质的定量分析,配备紫外检测器和自动进样器,保证样品分析的精度与重复性[1]。激光粒度分析采用Malvern Mastersizer 3000(英国马尔文公司),测量范围0.01~3500μm,用于分析不溶性杂质颗粒的粒径分布,并进行统计分析。扫描电子显微镜采用Hitachi SU8010(日本日立公司),用于观察杂质颗粒的形态与表面特征,确保图像分辨率和颗粒结构清晰可见。本研究使用了Eppendorf 5810R高速离心机(德国艾本德公司)进行样品分离,并采用Branson 5800超声波清洗仪(美国布兰森公司)对样品进行预处理[2]。所有设备均通过生产厂商的质量认证,确保实验结果的可靠性。
1.3 实验条件与实验过程
1.3.1实验环境设置实验在标准的实验室环境中进行,温度控制在20~25℃之间,湿度保持在45%~55%。该环境条件有助于确保仪器设备的稳定性,并减少外部环境对样品和数据的影响。实验前,所有溶剂和样品均在相同环境下保存,避免了温度变化可能带来的误差[3]。此外,实验室内所有设备,包括HPLC系统、激光粒度仪和扫描电子显微镜,均在相对稳定的环境条件下运行,以确保结果的精确性与一致性[4]。
1.3.2高效液相色谱法
在分析过程中,每个样品至少重复测量三次,以确保数据的准确性和重复性[5]。所有结果均通过标准曲线与已知浓度的标准品进行比较,从而获得不溶性杂质的准确含量。
1.3.3激光粒度分析
每次实验前,先对激光粒度仪进行校准,确保设备的测量精度。样品需通过液体分散,避免颗粒团聚[6]。激光粒度仪的测量范围从0.1~1000μm,可以准确获取样品中杂质颗粒的粒径分布情况。每个样品测量至少进行三次[7]。通过粒度分析,不仅能够判断杂质颗粒的大小,还能分析其对药物释放度和制剂稳定性的潜在影响。
1.3.4扫描电子显微镜分析
为避免表面电荷积聚影响成像质量,所有样品在测试前均经过喷金处理。SEM的加速电压为15 k V,工作距离为10 mm。通过对颗粒表面形态的观察,进一步探讨杂质对药物溶出、稳定性以及最终产品质量的影响。SEM成像为分析颗粒的形态提供了直观依据,帮助理解杂质颗粒是否对制剂的物理化学性质产生负面影响[8-9]。
1.4 评估标准不溶性杂质含量应符合药典限量要求,超标则判定不合格。药物释放度测试参照药典溶出度方法,需在规定时间内达到标准比例。颗粒粒径分布要求控制在合理范围内,避免过大颗粒影响药物溶出和稳定性。稳定性评估通过存储条件下的质量变化,考察不溶性杂质对药物释放度及外观的影响[10]。
1.5 数据来源本研究数据包括实验室HPLC、激光粒度仪、SEM等设备生成的样品数据,经过多次校正以确保准确性。标准品数据来自药典及国际标准,用于验证实验结果的可靠性。数据分析采用SPSS统计软件,保证统计显著性。
Part.02结果与分析
2.1 不同批次口服固体制剂中不溶性杂质的含量
不溶性杂质的含量是影响口服固体制剂质量的重要指标。各批次制剂的杂质含量受原料、生产工艺及储存条件等多种因素的影响,杂质含量过高可能影响药物的稳定性、释放度甚至疗效。不同批次口服固体制剂中不溶性杂质的含量见图1,不同批次的杂质含量差异明显。批次A的杂质含量最低,仅为0.3%,符合质量标准,表明生产控制较好。批次B和C的杂质含量较高,分别为1.2%和2.0%,可能反映生产过程中杂质控制存在问题,影响药物稳定性与释放度。批次D的杂质含量为0.7%,较为适中,但仍需进一步优化以减少杂质。批次E的杂质含量为0.4%,接近批次A,质量控制稳定,但仍有改进空间。
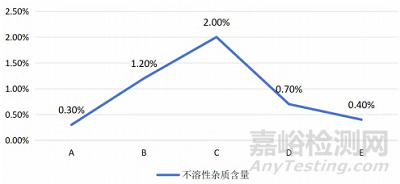
图 1 不同批次口服固体制剂中不溶性杂质的含量
2.2 不溶性杂质颗粒的粒径分布
不溶性杂质颗粒的粒径分布直接影响制剂的溶出特性与稳定性,粒径较大的颗粒可能影响药物的生物利用度,不同批次口服固体制剂中不溶性杂质颗粒的粒径分布见表1。激光粒度仪分析显示,不同批次制剂的杂质颗粒粒径分布差异明显。批次A颗粒较小,集中在0.1~5μm,平均3.2μm,最大8μm,影响较小。批次B颗粒较大,范围1~50μm,最大达50μm,可能显著影响药物释放。批次C的颗粒最大,范围5~100μm,最大98μm,可能影响溶出。批次D和E粒径较均匀,分布较小,预计对药物释放影响较低。
表 1 不同批次口服固体制剂中不溶性杂质颗粒的粒径分布
| 批次 | 颗粒粒径范围 /μm | 颗粒分布 /% | 平均粒径 /μm | 最大粒径 /μm |
|---|---|---|---|---|
| A | 0.1~5 | 45 | 3.2 | 8 |
| B | 1~50 | 60 | 8.6 | 50 |
| C | 5~100 | 30 | 15.2 | 98 |
| D | 0.5~30 | 55 | 5.3 | 30 |
| E | 0.1~10 | 40 | 2.5 | 10 |
2.3 不溶性杂质对药物释放度的影响
不溶性杂质,尤其是较大颗粒的杂质,可能导致药物释放速率的降低,从而影响药物的吸收和治疗效果,不同批次口服固体制剂中不溶性杂质对药物释放度的影响见表2,不溶性杂质含量与释放度呈负相关。批次A杂质含量较低(0.3%),30、60、120 min的释放度分别为45.2%、78.3%、98.1%。批次B和C杂质较高(1.2%、2.0%),释放度分别降至72.5%和65.4%,60 min后差异明显,可能影响药效。批次D和E杂质含量较低,释放度较好,但批次D在60 min后略低,可能受杂质粒径分布或形态影响。
表 2 不同批次口服固体制剂中不溶性杂质对药物释放度的影响
| 批次 | 药物释放度 (30 min) | 药物释放度 (60 min) | 药物释放度 (120 min) | 不溶性杂质含量 /% |
|---|---|---|---|---|
| A | 45.2% | 78.3% | 98.1% | 0.3 |
| B | 39.8% | 72.5% | 90.7% | 1.2 |
| C | 33.5% | 65.4% | 85.3% | 2.0 |
| D | 41.3% | 76.1% | 95.0% | 0.7 |
| E | 47.1% | 80.6% | 98.5% | 0.4 |
2.4 不溶性杂质对制剂稳定性的影响
不同存储条件下的稳定性测试显示,不溶性杂质对制剂的长期稳定性和外观质量具有明显影响(见表3)。批次A在室温存储条件下,不溶性杂质含量仅增长了0.3%,药物释放度变化较小,仅为1.5%,且外观无明显变化,说明该批次稳定性较好。批次B在高温高湿条件下存储,不溶性杂质增加了1.1%,药物释放度变化为3.4%,并出现了轻微结块现象,表明较高的杂质含量和存储条件的影响导致稳定性下降。批次C与批次A类似,室温存储条件下杂质增长较少,外观变化不明显,但药物释放度略有变化。批次D在高温高湿条件下,不仅杂质含量增加了1.3%,且药物释放度下降显著,结块现象明显,说明该批次的不溶性杂质对稳定性产生了较大负面影响。批次E表现与批次A相似,存储条件良好,杂质增长较少,药物释放度变化较小,稳定性较好。
表 3 不同存储条件下不溶性杂质对制剂稳定性的影响
| 批次 | 存储条件 | 不溶性杂质增加 /% | 药物释放度变化 /% | 外观变化 |
|---|---|---|---|---|
| A | 室温 (25 ℃) | 0.3 | 1.5 | 无明显变化 |
| B | 高温高湿 (40 ℃, 75% RH) | 1.1 | 3.4 | 轻微结块 |
| C | 室温 (25 ℃) | 0.5 | 2.0 | 无明显变化 |
| D | 高温高湿 (40 ℃, 75% RH) | 1.3 | 4.2 | 结块明显 |
| E | 室温 (25 ℃) | 0.4 | 1.2 | 无明显变化 |
Part.03讨论与结论
不溶性杂质对口服固体制剂的质量与稳定性具有显著影响,制剂中杂质含量与其药物释放度、长期稳定性以及外观质量密切相关。本研究通过不同批次的分析,揭示了不溶性杂质在制剂中的重要作用,并探讨了其对制剂性能的影响。
不溶性杂质的粒径和含量是影响药物释放度的关键因素。在本研究中,不同批次中杂质的粒径和含量差异显著,粒径较大的杂质颗粒往往会导致药物释放度的下降,在批次C中,较大颗粒的杂质对药物的释放速率产生了不利影响。高含量的不溶性杂质不仅会延缓药物的释放,还可能在储存过程中对制剂的稳定性产生负面效应。批次B和批次C的杂质含量较高,释放度降低,稳定性也表现较差。
针对不溶性杂质对制剂稳定性的影响,结果显示在不同存储条件下,杂质的增加导致了制剂外观的变化,尤其是在高温高湿条件下,杂质的积聚加速了结块现象,严重影响药物的生物利用度。批次D和B在高温高湿环境下的表现最为突出,结块现象对其后期的使用造成了潜在的风险。批次A和E在常温下储存时,杂质增加较少,稳定性表现较好。
综上所述,本研究验证了不溶性杂质对口服固体制剂质量的深远影响。未来的生产过程中,应进一步优化原料选择与生产工艺,严格控制不溶性杂质的含量,保证制剂的药效和稳定性。通过严格的质量控制,可以有效减少杂质积累,从而提高制剂的质量与市场竞争力。
参考文献
[1] 朱家乐, 刘清梁, 王莉. 口服固体制剂连续制造的工艺技术研究进展 [J]. 流程工业, 2024, (06): 44-47.
[2] 刘霏霏, 闫方, 宋晓, 等. 循环系统药物口服固体制剂仿制药生物等效性研究考虑要点 [J]. 中国临床药理学杂志, 2024, 40(05): 778-784.
[3] 徐晓宏, 李飞, 付盟, 等. 基于审评角度对口服固体制剂研发中粉体学性质控制的一般考虑 [J]. 中国医药工业杂志, 2023, 54(12): 1781-1788.
[4] 何驰宇, 朱雪萍, 胡玉玺. 口服固体制剂粒度和粒度分布相关问题探讨 [J]. 药学研究, 2023, 42(11): 884-890.
[5] 徐晓宏, 李飞, 付盟, 等. 口服固体制剂注册型研发中粉体学研究的一般考虑 [J]. 中国医药工业杂志, 2023, (11).
[6] 孟永刚, 侯鹏, 王亚敏. 口服固体制剂经鼻胃管给药体外评价试验的一般考量 [J]. 中国新药杂志, 2024, 33(07):651-653.
[7] 罗林秀, 管天冰, 罗安琪, 等. 口服固体制剂颗粒的离散元建模与破碎行为分析 [J]. 药学学报, 2024, 59(04):1057-1066.
[8] 李翠荣, 尚金燕, 张丽. 口服固体制剂技术在剂型药物制作中的进展研究 [J]. 北方药学, 2024, 21(01): 193-196.
[9] 魏赫, 李雪梅. 口服固体制剂注册申报工艺验证常见问题考量 [J]. 中国药学杂志, 2023, 58(22): 2098-2102.
[10] 王燕敏. 化学药口服固体制剂变更原料药供应商研究流程 [J]. 中国药业, 2023, 32(21): 73-79.

来源:Internet