
嘉峪检测网 2025-06-20 20:15
导读:本文对医疗器械MDSAP认证进行了详细介绍,见下文。
一、MDSAP是什么?
MDSAP是Medical Device Single Audit Program的首字母缩写,中文译为“医疗器械单一审核程序”,MDSAP认证项目是由美国FDA、澳大利亚TGA、巴西ANVISA、加拿大HC、日本MHLW/PMDA五国的医疗器械监管机构(Regulatory Authorities,简称RAs)共同认可并加入的质量管理体系审核程序。
MDSAP要求由五国医疗器械监管机构(RAs)联合认可的审核组织(Auditing Organizations,简称AOs))对医疗器械制造商进行质量管理体系审核,通过后由AOs颁发的MDSAP证书。AOs需将审核结果共享给参与该项目的监管机构(RAs),以支持其上市前审批和上市后监管工作。
获得MDSAP证书仅表明医疗器械制造商在质量管理体系符合参与国的法规要求,而非证明其产品的安全性和有效性已直接满足相关法规标准。
二、MDSAP认证介绍
01MDSAP定义
医疗器械单一审核程序(MDSAP)的总目标是开发、管理、监督一项单一审核程序,这一程序将允许对由MDSAP认可的审核组织对医疗器械制造商进行一次法规审核来满足多国法规在质量管理体系方面的要求。
02MDSAP项目背景
随着医疗器械国际贸易的不断发展,不同国家和地区各自的医疗器械质量管理体系审核给企业带来了沉重的负担。为了简化审核流程,提高审核效率,同时确保医疗器械的安全性、有效性和质量,MDSAP应运而生。它是由国际医疗器械监管者论坛(IMDRF)的成员共同推动创建的,旨在通过一次审核满足多个国家和地区法规在质量管理体系方面的要求,大大减少了企业在不同国家分别接受审核的时间和资源成本。
03MDSAP目前参与成员国
◆ 美国食品药品管理局(FDA) U.S. Food and Drug Administraion
◆ 澳大利亚药品管理局(TGA) Therapeutic Goods Administration of Australia
◆ 巴西国家卫生监督局(ANVISA) Brazil’s Agência Nacional de Vigilância Sanitária
◆ 加拿大卫生部(HC)Health Canada
◆ 日本厚生劳动省/医药医疗器械管理局(MHLW/PMDA) Japan’s Ministry of Health, Labour and Welfare / Pharmaceuticals and Medical Devices Agency
MDSAP正式观察员MDSAP Official Observer:
◆ 欧盟委员会 European Union (EU)
◆ WHO IVD资格预审项目组
◆ 新加坡卫生科学局(HSA)
◆ 英国药品和健康产品管理局(MHRA)
04MDSAP相关国家认可程度
01美国:
◆ 美国FDA接受MDSAP运行审核报告来取代FDA常规监督检查“routine surveillance inspection”(通常每2年1次)
◆ FDA不会接受MDSAP报告作为跟踪检查“Follow-up Inspections”或有因检查“For-cause Inspections”。
◆ MDSAP审核报告不适用任何上市前许可PMA审查及安全性评估程序。
◆ 对ERPC电子产品辐射控制法案的要求将由FDA继续执行检查。
◆ 如果被审核工厂只是美国体系下的一部分,FDA可能仍然维持日常检查或有因初始检查。
◆ 在FDA例行检查公布之前,受审核组织必须已经签好MDSAP审核合同,否则FDA仍旧可能进行现场检查。发证机构将提前告知主管当局要进行MDSAP审核。
02加拿大:
◆ 加拿大卫生部使用MDSAP证书作为医疗器械法规第32(2)(f)条、32(3)(j)及32(4)(p)的符合性证据。
◆ MDSAP证书取代了CMDCAS证书。
◆ 2015年12月4日起,加拿大卫生部公布了MDSAP转移计划,从2019年1月1日起,在II/III/IV类产品上市许可审查阶段,只有MDSAP证书能够被HC接受。
03巴西:
◆ 根据RDC 15/2014和RE 2.347/2015的定义,ANVISA可使用MDSAP审核结果来代替ANVISA的上市准入审核并签发ANVISA的GMP证书,该证书用于III类和IV类医疗器械准入巴西市场。
◆ MDSAP审核可以加速ANVISA的GMP认证流程。
◆ ANVISA还可以使用MDSAP审核报告以每两年更新ANVISA的GMP证书,作为ANVISA全面检查的替代方案。如果制造商在先前的ANVISA审核中未通过,ANVISA将不会使用MDSAP审核报告来替代GMP审核。
◆ 2024年3月20日,ANVISA官方发布了RDC 850/2024号决议,正式将通过MDSAP授予医疗器械制造商的BGMP证书的有效期从两年延长至四年。
04澳大利亚:
◆ TGA将视MDSAP审核报告为:
-- 制造商能否证明符合澳大利亚符合性评估程序;
-- 是否能发放或维持TGA符合性评估证书
◆ 在某些情况下,制造商将免除日常TGA的审核;
◆ TGA接受MDSAP证书作为符合ISO13485的证据。
◆ 对于由TGA考虑的MDSAP证书和审核报告必须涵盖澳大利亚的监管要求,证书必须表明制造商已经过评估,并符合Therapeutic Goods (Medical Devices) Regulations 2002的法规要求。
05日本:
◆ 从2016年6月起,MHLW和PMDA在日本法规框架下,接受使用MDSAP审核报告用于上市前准入和上市后定期监督。
◆ PMDA允许上市许可持有人(MAH)用MDSAP审核报告报告替代检查所需的相当一部分文件,以代替J-QMS现场审核,减轻医疗器械制造商的负担,当提交MDSAP审核报告时,PMDA可以执行非现场检查代替现场检查或减少非现场检查的文件。
◆ 在审核MDSAP报告后确定有必要时,PMDA可以进行现场检查或要求制造商提供额外的质量管理体系文件。
◆ 当涉及生产动物源类、人源类产品或放射性IVD产品,MDSAP证书将不能被PMDA接受。
05MDSAP遵循的法规要求或审核依据
授权和认可的审核组织(AOs)根据参与监管机构(RA)制定的文件执行MDSAP审核。一些相关政策和程序的引入确保了项目的一致性,MDSAP审核主要参考文件:
MDSAP AU P0002审核手册要求
◆ MDSAP AU P0002 Audit Approach
MDSAP AU P0008 审核时间要求
◆ MDSAP AU P0008.008: Audit Time Determination Procedure
◆ MDSAP AU F0008.2.002 Audit Duration Calculation Form (Audit Model 2017)
MDSAP AU P0019 审核报告要求
◆ MDSAP AU P0019.004 Medical Device Regulatory Audit Reports Policy
◆ MDSAP AU F0019.1.008 Medical Device Regulatory Audit Report
◆ MDSAP AU F0019.2.011 NC Grading and Exchange Form
◆ MDSAP AU G0019.3.007 Medical Device Regulatory Audit Report Form Guidelines
◆ MDSAP AU G0019.4.004 Guidelines NC Grading Exchange Form
MDSAP AU P0026 认证证书要求
◆ MDSAP AU P0026: Certificate Document Requirements
◆ MDSAP G0026.1.004 Surveillance Audit Confirmation Notification Process
MDSAP AU P0027 审核后活动与时限要求
◆ MDSAP AU P0027.007 Post Audit Activities and Timeline Policy
其他审核参考文件请见
◆ MDSAP Audit Procedures and Forms | FDA
06MDSAP审核模型
MDSAP每个审核过程都有一些审核任务组成,审核模型将直接指导审核员来收集每个任务对应的证据,每个任务对应ISO13485的条款和特定参与国的法规要求,风险管理贯穿在整个审核过程当中。
MDSAP审核基于四个主要过程以风险管理为导向的过程方法审核思路:
◆ 管理过程(11个审核任务)
◆ 测量分析和改进过程(16个审核任务)
◆ 设计和开发过程(17个审核任务)
◆ 生产和服务过程控制(29个审核任务)
以及三个支持过程:
◆ 医疗器械上市许可和工厂注册过程(3个审核任务)
◆ 医疗器械不良事件和忠告性通知报告(2个审核任务)
◆ 采购过程(12个审核任务)
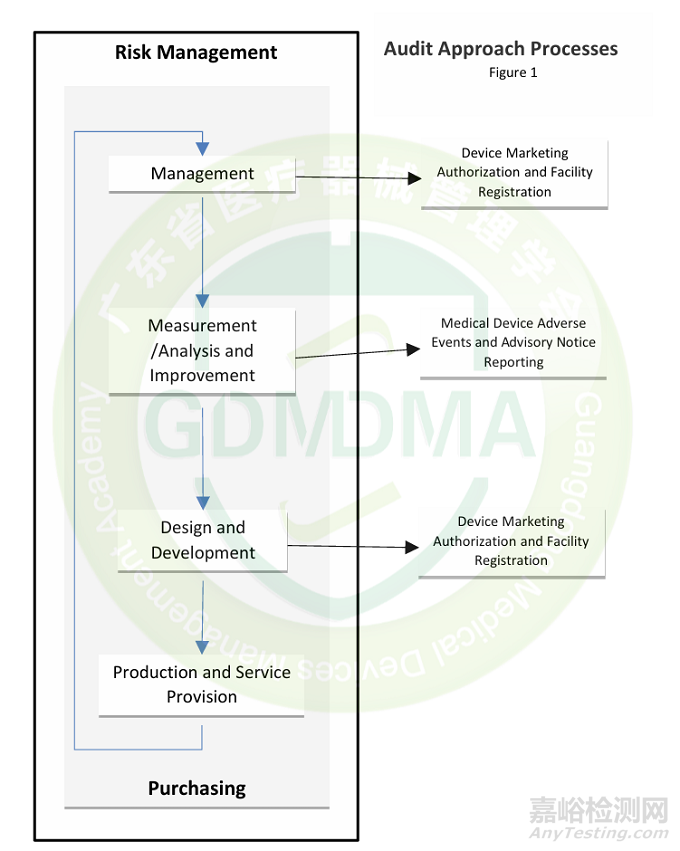
注:虽然MDSAP过程(process)有规定的审核顺序,但审核员可以在给定过程中以任何顺序审核任务(task),以实现高效和有效的审核。
07MDSAP认证机构
以下列出了提交医疗器械单一审核程序(MDSAP)申请的审核组织(AOs)清单,同时显示了这些审核组织(AOs)的申请状态,MDSAP审核的授权以及认可情况。
Auditing Organization Availability to Conduct MDSAP Audits | FDA
目前可以进行MDSAP审核的审核组织(AOs)可点击如上链接进行查询。
举例:TÜV SÜD、TÜV Rheinland、BSI、DEKRA、DNV、UL等。
08DSAP审核类型
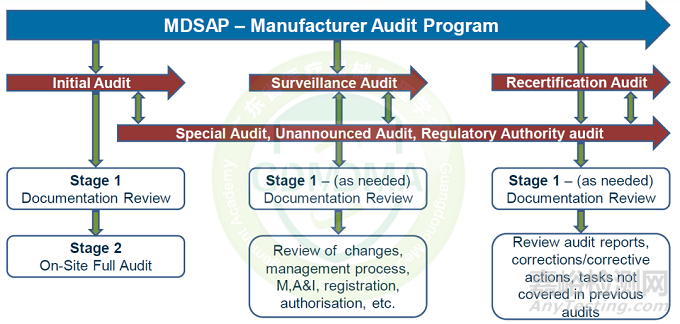
MDSAP按照审核类型,主要分为初次审核(一阶段审核、二阶段审核)、监督审核、再认证审核、不通知审核、特殊审核和主管当局审核。MDSAP审核周期为3年,在初次审核之后第1、2年每年须接受一次监督审核,第3年须进行再认证审核。初次审核和再认证审核均为全面审核,监督审核为部分审核。
1)初次审核:主要包括一阶段审核和二阶段审核。一阶段审核包括文件审查、评估二阶段审核的准备情况等;二阶段审核主要对QMS执行情况和有效性进行评估。一阶段审核的部分内容(如文件审查)可在初始认证的医疗器械制造商所在地以外的地点进行。但二阶段审核应在证书记录的所有场地进行现场审核。因此,任何与医疗器械制造商QMS相关但非现场审核的场所都不应记录在证书上。
2)监督审核:监督审核不需要一阶段审核,除非自上次审核以来发生了重大变更。一个审核周期内应有两次监督审核,审核周期中的每个监督审核不需要涵盖所有MDSAP要求。然而,每次监督审核任务Tasks的最低限度需参考MDSAP AU P0008.008: Audit Time Determination Procedure的Appendix 1 – Surveillance Audits,如:
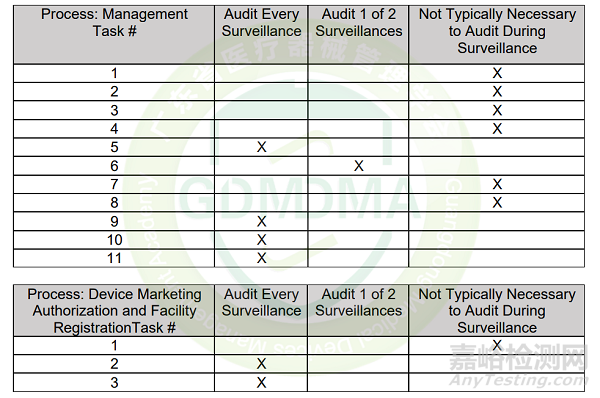
3)再认证审核:再认证审核不需要一阶段审核,除非自上次审核以来发生了重大变更。再认证审核应在证书记录的所有场地进行现场审核。因此,任何与医疗器械制造商QMS相关但非现场审核的场所都不应记录在证书上。
4) 特殊审核:特殊审核不是计划审计周期的一部分。特殊审核仅在必要时使用,并应侧重于医疗器械组织质量管理体系的特定要素。
特殊审核用来处理以下情况:
◆ 医疗器械制造商需要扩大认证范围的审核;
◆ MDSAP认可的审计机构缺乏监督。例如,由于审核时间不足,审核组组成不合理等。
◆ 跟进上市后的具体问题。例如,对于潜在的重大投诉。
◆ 跟进先前MDSAP审核的重要发现
◆ 应MDSAP参与监管机构(RA)的要求(基于特定任务)
◆ 根据监管机构(RA)或审核组织(AOs)的政策要求对供应商进行审核。
执行特别审核的审核组织AOs应在审计最后一天起15天内向认可监管机构(RA)提交审计报告。
5)不通知审核:不通知审核也属于特殊审核中的一种,如我们通常所说的飞行检查。MDSAP参与监管机构(RA)要求审核组织AOs在发现高等级不符合项(high grade non-conformities)的情况下进行不通知审核,以确保医疗器械制造商质量管理体系运行的有效性。
监管机构(RA)也可能在任何时候开展对医疗器械制造商的直接审核。例如:
◆ 监管机构(RA)获得的关于制造商各方面负面信息导致的审核;
◆ 监管机构(RA)对以前审核结果的后续审核;
◆ 确认MDSAP认可的审核组织(AOs)有效实施MDSAP要求。
09MDSAP对产品技术文件的要求
下表总结了MDSAP审核员将用于审查构成技术文档的信息的任务。
| Information | Audit Approach: Process, Task# |
|---|---|
| Medical device general description, including variants and accessories | Design and Development, task #5, 7 |
| Evidence of compliance with specified regulatory requirements for products or processes.9 | Design and Development, task #5, 7 |
| Evidence of inclusion of feedback into risk management for monitoring and maintaining the product requirements as well as product realization or improvement processes | |
| Information that confirms that design and development outputs for the product are traceable to, and satisfy, design input requirements | Design and Development, task #7 |
| Intended use, and indication of use, of the medical device | Design and Development, task #5, 7, 10, 11 |
| Labelling, (i.e., information that accompanies a medical device that is located on the device, its packaging, the instructions for use and in promotional material) | Design and Development, task #1, 7, 8, 16 |
| Confirmation that the product is a medical device |
Device Marketing Authorization and Facility Registration, task #1 Design and Development, task #5 |
| Classification |
Device Marketing Authorization and Facility Registration, task #1 Design and Development, task #5 |
| Risk management file | Design and Development, task #8 |
| Pre-clinical data (studies in animal models, testing to support compliance with relevant standards, technical performance tests etc.) | Design and Development, task #10 |
| Clinical evidence | Design and Development, task #11 |
| Manufacturing processes |
Design and Development, task #7, 16 Production and Service Controls, task #3, 16 |
| Process validation |
Design and Development, task #16 Production and Service Controls, task #7, 8, 9 |
| Evidence of compliance with specified regulatory requirements for marketing authorization. | Device Marketing Authorization and Facility Registration, task #1 |
| Declaration of conformity | Device Marketing Authorization and Facility Registration, task #1 |
Note: this table may not exhaustively cover all information expected under all jurisdictions.
监管机构(RAs)的期望:
每个参与的监管机构(RAs)对技术文件的审查和在审核时评估该技术文件的充分性有不同的要求。其中,澳大利亚治疗商品管理局(TGA) 的审核要求最为严格。
10MDSAP审核不合格项分级
MDSAP审核中不使用严重性和轻微项简单不合格分级,而引入了标准分级系统,按照下图分级矩阵进行分级,结合分级上升规则,最终不合格分级共分5级(1-5)。

◆ 非直接QMS影响Indirect QMS Impact
ISO 13485条款4.1至6.3(分级1-2)
◆ 直接QMS影响Direct QMS impact
ISO 13485条款6.4至8.5(分级3-4)
◆ 以下情况需要立即通知发证机构,且如果存在1项5级或超过2项4级不合格项将会触发飞行检查(一般在9个月之内)
1)1项或多项5级不合格;
2)超过2项4级不合格;
3)存在公众健康威胁;
4)任何欺诈行为或伪造医疗器械产品。
MDSAP审核不合格项回复时间要求:
1)所有不合格项需要在审核日起15天内回复原因,纠正及纠正措施计划;
2)对于1-3级不合格项,支持不合格关闭的证据在下次例行审核中进行跟踪;
3)对于4或5级的任意不合格项,需要在审核日起30天内提供有效的纠正措施执行的证据。
11MDSAP认证申请注意事项
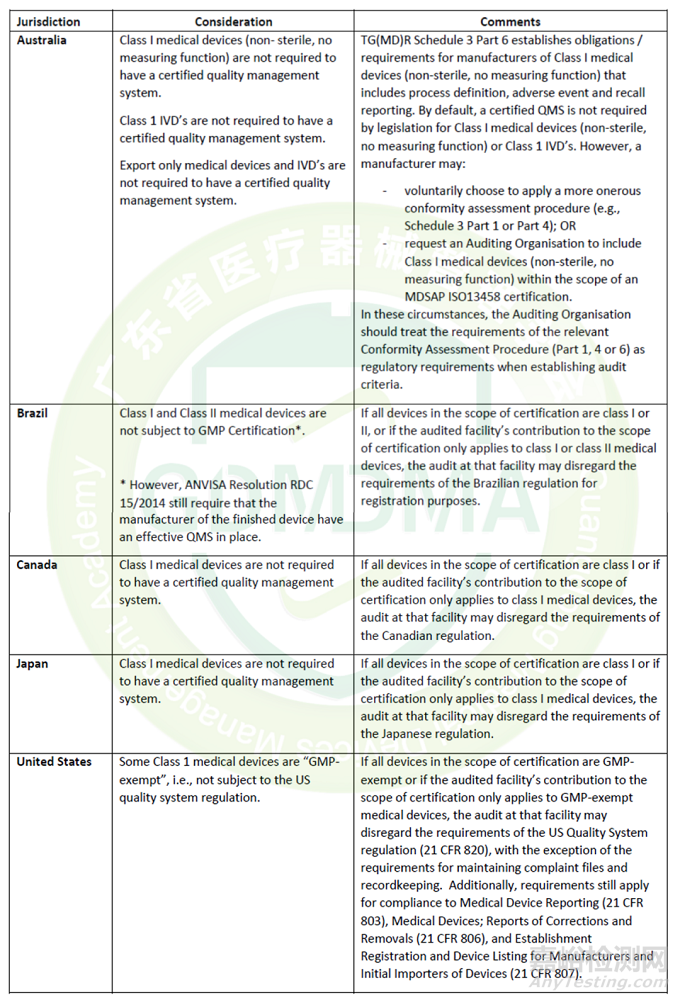
1)如果企业申请MDSAP之前未向MDSAP的5个国家出口过任何产品,认证时申请的国家数量可自行根据业务发展需要选择。
如果企业有向其中的一个或多个国家进行过出口或在当地注册,则该国家必须包含在申请范围内。以下情况除外。
2)MDSAP认证机构可以为医疗器械制造商颁发尚未获得上市许可地区的MDSAP证书,考虑到产品准入市场可能需要时间,此类认证可以延长三年时间,如三年期满,制造商仍未任何产品获得或向该区域申请上市许可,则认证机构会建议该地区从现有证书中移除,直至制造商能够证明相关要求的执行情况及有效性。

来源:广东省医疗器械管理学会