
嘉峪检测网 2025-06-26 21:52
导读:本文详细介绍了药包材要做微生物检测吗等内容,详见下文。
引子:前不久药典委公布了《药包材微生物检测指导原则征求意见稿》,这一行动标示着药包材的微生物检测快要进入药典了。看了之后,感慨颇多。后台也接到了一些朋友的疑问。一些困惑和思考,抛砖迎玉,欢迎大家留言表达你的意见。
1国际上没有要求?

文章的讨论部分:“目前,美国食品药品监督管理局(FDA) 、欧盟药品监管局(EMA)、美国药典、欧洲药典、日本药局方等指南或标准中均未明确提出药包材微生物检验的方法及标准,虽然世界卫生组织(WHO)第 902号技术报告附录 9 药品包装指南中明确要求进行药包材的微生物检验,但其引用的是 EMA 1998年的文件,而在 EMA 2005 年的文件中并未要求药包材的微生物检验。国际上对药包材的微生物标准几乎没有明确要求。”
这些标准中没有要求是不是代表国外的药包材不要微生物指标监控呢?
2国外有要求
无菌药品:


FDA指南文件无菌产品的容器和密封件对无菌有明确要求。但是更多内容是制剂企业在收到无菌产品药包材之后的清洗、漂洗和灭菌过程的验证,还有包装的无菌屏障性能。
如果对容器和密封件进行灭菌和/或除热原进行外包,也应符合 cGMP 要求。成品剂型制造商应审查和评估外包商的验证方案和最终验证报告。
非无菌药品:


非无菌药物剂型的初级包装的设计、生产和加工方式不得改变药物的安全性、特性、质量和纯度。对于某些产品,例如液体制剂或吸入剂,需要解决通过初级包装引入的微生物污染。在初级包装而不是药品本身中使用抗菌活性物质可能是一个有吸引力的选择。本章介绍了一种科学方法,其中根据应用途径和与药物接触的初级包装表面来定义标准。许多初级包装材料可以使用膜过滤法进行测试。
综合以上无菌药品和非无菌药品的相关内容可以看出,国外对药包材的微生物水平不是没有要求,只是重点并不在药包材本身微生物的检测。
3药包材微生物检测本身
按照征求意见稿,药包材的微生物检测主要包括下表的内容。
表1. 药包材成品检测项目、指标要求、检测频次
| 包材类型 | 检测项目 | 指标要求 | 检测频次 |
|---|---|---|---|
| 无菌 | 无菌检查 | 无菌生长 | 逐批检查 |
| 非无菌 | 无菌药品用 | 生物负载测定(由供需双方确定) | 由供需双方在企业标准或质量协议中规定,应保证每批产品的生物负载均符合可接受水平规定。 |
| 非无菌药品用 | 微生物限度检查 | 由供需双方在企业标准或质量协议中规定,应保证每批产品的微生物限度均符合限度标准规定。 |
但是,由于药包材的特殊性,相关测试的前处理与药品可以说完全不同,倒是与医疗器械的处理方式雷同。所以在征求意见稿中直接标明参考了GB/T 19973.1《医疗保健产品灭菌微生物学方法第 1 部分:产品上微生物总数的确定》,其实就是ISO11737-1标准内容。
无菌药包材:
第一种情况无菌药包材的无菌检查其实价值不大,优先选择直接接种法。由于行制和大小问题,还要进行拆散和切碎处理,这些人为操作加大了人为污染的可能。次选择是洗脱法,因为药包材也只是要求内部无菌,这时候的洗脱方式和洗脱液就更加多种多样,洗脱效率参差不齐。方法选择时也可以参考一下GB/T 14233.2《医用输液、输血、注射器具检验方法》,里面有一些不同类型器械产品无菌检查方法的举例,可以参考。
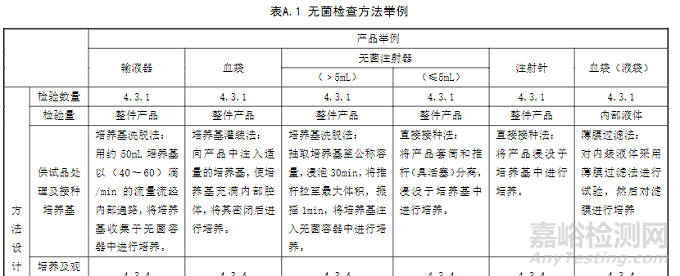
但是,无菌药包材更应该关注的是药包材的灭菌验证。如果这是由药包材供应商执行的,成品制剂企业更应该是加大关注力度。
非无菌药包材:
第二种和第三种情况的非无菌药包材检测的难点就是检测方法的检出率的问题。参考GB/T 19973.1,可以有很多方法的选择,比如擦拭、冲洗、振摇、超声波等等,以及不同类型冲洗液的选择。但是不得不承认,最后的效率还是一般。生物负载测定时如何设计多次验证设定科学的回收率进行后续补偿是主要问题。微生物限度检查中的控制菌检查也会遇到低限度水平的药包材检出率低的问题,如下图。

表5 垫片检出率结果
| 浓度 | 供试液增留法检出率(阳性样本数/总样本数) | 直接投入增留法检出率(阳性样本数/总样本数) |
|---|---|---|
| 接种浓度一 | 100%(10/10) | 100%(10/10) |
| 接种浓度二 | 100%(10/10) | 100%(10/10) |
| 接种浓度三 | 80%(8/10) | 90%(9/10) |
| 接种浓度四 | 46.7%(14/30) | 55%(22/40) |
在这个研究中,直接投入增菌法检出率略高于供试液增菌法,但是直接投入法操作简单,步骤较少,减少了外来污染。
因为测试效率不高,所用方法的选择和接收指标就要药包材供应商和成品制剂企业之间协商确定。对于非无菌药包材来说,更重要的时产品质量的稳定性,就包括微生物水平。

来源:Internet