
嘉峪检测网 2025-09-05 19:51
导读:本次研究依据保证药品安全性和有效性的原则对市售的尼可地尔片开展按法定标准检验和探索性研究,对质量标准项目设置的全面性、合理性以及制剂中的有关物质来源、影响因素,溶出曲线情况和样品含量等进行考察,根据研究结果对该品种相关质量标准存在的问题和品种质量情况进行了深入的分析,并提出相关评价和建议,以期为药品监管部门和相关生产企业提供参考。
摘要
目的:分析与评价尼可地尔片的质量标准可行性及质量状况。
方法:采用法定检验标准对88批尼可地尔片进行检验和统计分析,并采用探索性研究的方式对国内企业生产的尼可地尔片的有关物质、溶出曲线、含量均匀度和含量测定等项目进行了考察与评价。
结果:按法定检验标准检验了共88批尼可地尔片样品,合格率100%。通过探索性研究发现相关法定检验标准项目设置不完善,部分方法专属性较差,存在不能准确测定真实含量的问题;部分辅料、生产工艺及包装易导致样品水分增加,影响稳定性。
结论:收集到的尼可地尔片与原研制剂存在差距,建议相关企业选择适宜的辅料加强原辅料的水分控制及工艺过程中水分、温度等参数控制并改进包装,以降低杂质产生量;采用区分度高的溶出度控制方法进行质量控制,进行仿制药质量与疗效一致性评价,提高产品质量。目前相关质量标准存在缺陷,建议针对鉴别、有关物质、溶出度、含量均匀度和含量测定项目进行完善。
关键词
尼可地尔片;质量分析;药品监管;仿制药一致性评价
尼可地尔(Nicorandil),化学名为N-(2-羟乙基)烟酰胺硝酸酯,属硝酸酯类化合物,是一种ATP敏感性钾通道开放剂,一般由N-(2-羟乙基)烟酰胺经硝酸酯化得到,或由氨基乙醇硝酸酯,再经酰化反应生成。该品种由日本中外制药株式会社于1978年首先试制成功,临床上主要用于治疗冠心病,心绞痛[1-6]。尼可地尔的国产仿制药最早由江苏省徐州医药科学研究所与丰县制药厂协作研制,1986年1月由江苏省卫生厅批准丰县制药厂生产原料和片剂并上市[7]。为更好评价市售尼可地尔片的质量,并对相关质量标准和影响药品质量的关键参数进行分析,本次研究依据保证药品安全性和有效性的原则对市售的尼可地尔片开展按法定标准检验和探索性研究,对质量标准项目设置的全面性、合理性以及制剂中的有关物质来源、影响因素,溶出曲线情况和样品含量等进行考察,根据研究结果对该品种相关质量标准存在的问题和品种质量情况进行了深入的分析,并提出相关评价和建议,以期为药品监管部门和相关生产企业提供参考。
1仪器与试药
1.1 仪器
LC-20A高效液相色谱仪(日本岛津公司),1200高效液相色谱仪(美国Agilent公司),2690高效液相色谱仪(美国Waters公司),SOTAX自动溶出仪(瑞士SOTAX公司),RCZ-8M自动溶出仪(天津天大天发科技有限公司),ZB-1E智能崩解仪(天津天大天发科技有限公司),TU-1901紫外可见分光光度计(北京普析通用仪器有限公司),QE高分辨液质联用仪(美国Thermo公司),AVANCEⅢ400兆核磁共振谱仪(德国Bruker公司),µDissTM型光纤药物溶解性测定仪和µFluxTM型光纤药物渗透性测定仪(美国Pion公司),UltimaⅣ型粉末X射线衍射仪(日本Rigaku公司),Mastersizer2000激光衍射粒度分布仪(英国Malvern公司),Titrando890水分测定仪(瑞士Metrohm公司),KB240恒温恒湿箱(德国Binder公司),XA205电子天平(美国Mettler Toledo公司)。三氟乙酸、三乙胺、四氢呋喃等流动相用试剂均为色谱纯,水为纯化水。尼可地尔对照品由中国食品药品检定研究院提供,尼可地尔片样品均来源于国家药品计划抽验。
1.2 样品
通过查询国家药品监督管理局网站数据库,尼可地尔片国内共有56家生产企业56个批准文号,但仅有部分企业实际生产。本次研究共获得两家生产企业共88批样品,经调研,A企业采用湿法制粒压片法,B企业和原研企业均采用粉末直接压片法进行生产。获得的88批样品覆盖我国18个省、自治区、直辖市,样品来源涉及生产企业、经营企业和医疗机构,其中生产企业1批(1.1%);医疗机构21批(23.9%);经营企业66批(75.0%)。样品覆盖地域范围大,来源分布广泛,涉及全国范围内药品生产、经营和使用的各个环节,具有评价意义。
相关产品的药品质量标准为WS-10001-(HD0151)-2002[8],英国药典亦有收载[9]。
2检验结果及质量分析
对获得的88批尼可地尔片样品按国家药品标准WS-10001-(HD-0151)-2002(以下简称国家药品标准)检验,检验项目包括性状、2项化学反应鉴别、溶出度、含量均匀度和含量测定,结果均符合规定,合格率为100%。A企业样品测定结果显示,不同地域同批号样品测定结果间有差异,B企业不同地域同批号样品测定结果间无明显差异,故地域因素会影响A企业药品质量,但未影响B企业药品质量。
2.1 鉴别
国家药品标准鉴别方法为取本品细粉适量(约相当于尼可地尔0.15g),加丙酮10mL,振摇使溶解,滤过,滤液置水浴上蒸干,取残渣照下述方法试验。化学鉴别(1):取残渣约0.1g,加水与氢氧化钠试液各5mL,溶解后,缓缓煮沸,即产生刺激性气味,继续加热至气味完全除去,加酚酞指示液1~2滴,用稀硫酸中和后,加硫酸铜试液2mL,缓缓析出淡蓝色沉淀,滤过,取沉淀灼烧,即产生吡啶臭气。经检验所有批次样品均呈正反应,合格率100%。化学鉴别(2):取残渣约10mg,置试管中,加水1mL与硫酸2mL,振摇使溶解,放冷,沿管壁缓缓加硫酸亚铁试液3mL,两液层接界面显棕色。经检验所有批次样品均呈正反应,合格率100%。
2.2 溶出度
国家药品标准中溶出度采用小杯法,盐酸(稀盐酸24→1000)溶液250mL为溶出介质,转速50rpm,30min取样,在261nm下用紫外E值法测定,限度为70%,经测定,88批样品均符合规定,合格率100%。A企业49批样品的溶出度均值最小值为86%,最大值为108%,平均值为100%。B企业39批样品的溶出度均值最小值为94%,最大值为106%,平均值为100%。结果表明,不同生产企业样品的平均溶出量中位值均在100%附近,但平均溶出量结果差异较大,结果分布较为分散。其中A企业样品最低与最高溶出度均值相差22%,最低与最高批内RSD相差10.15%,B企业样品最低与最高溶出度均值相差12%,最低与最高批内RSD相差7.26%,两家企业样品批间与批内溶出度差异均较大,其中A企业样品批间与批内溶出度差异相对更大。溶出度结果显示,虽然两家企业的样品均合格,但结果离散性均较大。
2.3 含量均匀度
与溶出度相似,国家药品标准中含量均匀度检查也为在261nm下用紫外E值法测定,限度为A+2.2S≤15.0。测定结果显示,两家企业A+2.2S平均结果差异不大。但比较不同企业样品的含量均匀度A值与S值,可以看出A企业各批样品间S值差异相对较小且集中,说明工艺相对稳定,但A值差异较大,显示其样品批间差异相对较大,说明投放原料不稳定或样品发生不同程度的降解;而B企业各批样品间的S值差异相对较大,显示其样品批内差异较大,说明其压片等工艺的稳定性有待提高,而A值差异相对较小,说明其质控相对稳定。综上所述,88批样品含量均匀度虽都合格,但A企业样品批间差异较大,而B企业批内差异较大,因此两家企业有必要加强生产工艺,使样品均一性更好。
2.4 含量测定
对获得的共88批次尼可地尔片按国家药品标准进行测定,检验结果均在限度90.0%~110.0%,均符合规定,合格率100%。结果显示,A企业样品含量测定均值略高于B企业样品,不同生产企业样品的含量测定结果差异较小,结果分布较集中。另外在对A企业同批号不同地域样品的检测中发现,其中41.3%的样品含量测定结果超出104%的情况,且同批不同地域样品含量测定差异极值大于6%。分析导致该结果的原因可能有三方面:①企业超100%投料;②样品含量测定专属性不佳,降解产物的紫外吸收大于主成分,干扰了测定结果;③包装隔离不足,导致环境湿度等条件对样品主成分产生了影响。综上,两家企业样品含量均值均高于100%,但同批号样品含量差异大,提示可能与企业制备工艺、包装以及储藏有关。
2.5 现行国家标准存在的问题
通过评估,现行国家标准不能全面分析国内尼可地尔片的产品质量,其主要问题有:①项目设置不完善,无有关物质检查项,缺乏对药品安全性进行有效控制的手段。②尼可地尔片鉴别项中有通过闻氮氧化物及吡啶的方式进行鉴别的步骤,对检验员身体健康有影响,且缺乏专属性较高的鉴别项目。③现行国家标准含量均匀度、含量测定均采用紫外E值法测定含量,专属性差,不能准确测定其真实含量。④现行国家标准方法采用小杯法,使用盐酸溶液作为溶出介质,30min取样,限度为70%,与原研制剂采用的大杯法有差异,不利于仿制药一致性评价工作的进行,且检测方法为紫外E值法,专属性差,不能准确测定溶出液中的主成分含量。
3探索性研究结果及质量分析
根据国家药品标准检验中发现的问题,围绕样品的安全性和有效性进行了探索性研究。
3.1 有关物质
因现行国家标准未收载有关物质检查项,且其含量测定等方法均为紫外E值法,专属性较差,因此对尼可地尔片的有关物质情况了解不深入。目前英国药典虽有收载采用高效液相色谱法(HPLC)的尼可地尔片有关物质检查项,2017年版本对方法进行了变更,但是其新旧方法均无法有效分离国内尼可地尔片的有关物质[10-12]。因此,本研究团队首先建立了一个能够对国内尼可地尔片样品进行有效分离的高效液相色谱有关物质检查方法对样品的有关物质进行测定[13]。采用高效液相色谱-质谱联用(LC-MS)、核磁共振等技术对样品中存在的杂质进行考察,明确杂质结构。通过ADMET Predictor软件预测毒性和细胞毒性试验考察杂质毒性,结果显示杂质毒性均较小,且均低于主成分尼可地尔。用建立的高效液相色谱有关物质检查方法对88批样品进行了检验,样品杂质含量较高,平均杂质总量为9.9%(图1)。其中A企业样品的杂质总量和各单个杂质量与离散值显著高于B企业样品,反映出样品相对质量较差。通过对原料药及原研制剂的测定,发现各企业的原料药杂质量均较低(杂质总量不超过0.1%)且原研制剂的杂质总量明显低于国内企业。由结果可得:①采用粉末直接压片样品(原研和B企业)的有关物质明显少于湿法制粒压片样品(A企业),说明水分控制是影响有关物质产生的关键因素。②同样采用粉末直接压片工艺生产的样品(原研与B企业),其有关物质的量差异依然很大,说明辅料及相关工艺也是影响产品稳定性的关键因素。③制剂中的杂质含量比原料药中明显增大,说明主成分易受处方、制剂工艺、储藏条件的影响。
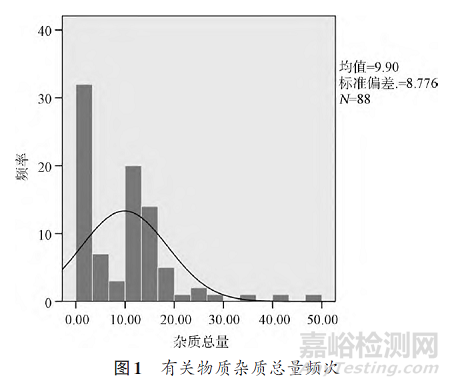
围绕以上问题开展了5个方面的延伸性探索性研究。①在对A企业采用的湿法制粒压片生产工艺研究中发现,样品在制粒到压片的过程中,每个步骤中杂质都有所增长,尤其是在制粒过程,杂质总量增长较快(3.1%),推测制粒中的湿热条件是导致杂质显著增加的关键因素,该推论在后续进行的探索性研究中得到了确证。此外,压片后片剂的杂质总量为3.8%,而获得的A企业共49批次样品有关物质杂质总量的均值明显更高,说明更多的杂质是在长期储藏过程中产生的。②在稳定性影响因素研究中发现在高温条件下原料药和制剂杂质都随时间延长显著增加,说明尼可地尔不能耐受高温。据调研,尼可地尔在40~70℃时会发生聚合反应不断生成多聚体[14],使杂质量不断增加。高湿条件下,各厂家原料药的杂质均有增加,而制剂吸湿明显,杂质增加量明显高于原料药,表明尼可地尔片对湿度较为敏感,其制剂存在原辅料相容性问题。在强光照射条件下,原料的杂质量有所增加,制剂相对于原料药更加稳定,可能因尼可地尔被辅料包裹,较少被光照直射有关。③通过对原研和国内企业样品的水分进行测定,结果显示,A企业的样品水分(均值7.9%)明显高于B企业(均值5.4%),而原研药水分均值仅为0.6%。通过相关性分析,样品的水分值与所有杂质的生成显著正相关(P<0.01),与影响因素试验高湿试验结果吻合,说明水分严重影响尼可地尔的稳定性。④在原辅料相容性研究中发现,引湿性较强的辅料如羧甲基淀粉钠、糊精等,均会导致杂质显著增加,说明该类型辅料存在着原辅料相容性问题,应尽量避免使用。⑤在包装影响研究中发现,在同样温度条件下,处于加干燥剂的包装内的样品杂质增加相比裸片更缓慢,说明高湿度环境会加快尼可地尔的降解速度。在长期的放置中铝塑板内的片剂的杂质量会与裸片趋于等同,单纯的铝塑板不能有效起到保护制剂的作用,相比之下双铝箔板隔绝效果更佳。此外,虽然同样采用了铝塑板(或双铝箔板)加铝箔袋内附干燥剂的包装形式,但原研制剂的杂质量可维持在相对较低的水平,而B企业样品的杂质量逐渐上升。提示两个企业的制剂可能存在着干燥剂效能方面的差异。通过干燥剂效能试验,结果表明,原研药所用的干燥剂吸湿性能为B企业的10倍以上,可以更有效地控制铝箔袋中湿度。
3.2 溶出度
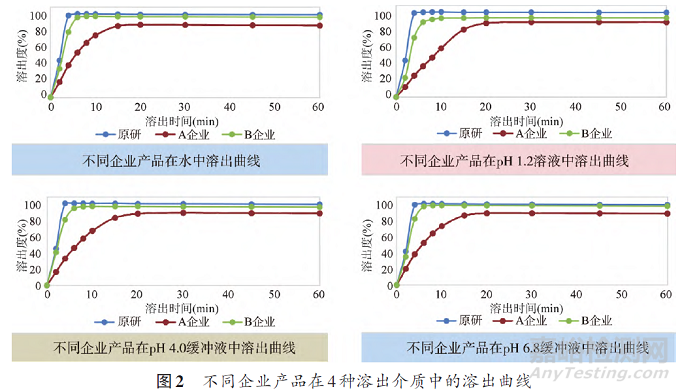
口服制剂在人体中的生物利用度是评价药物有效性的重要指标,其中溶出度作为体外手段体现口服制剂的生物有效性,可通过考察不同pH值溶出介质中的溶出曲线来评价制剂的生物有效性[15,16]。参照日本橙皮书条件[17],按照溶出度与释放度测定法(《中国药典》2020年版通则0931第二法)[18],分别以水、pH1.2、pH4.0、pH6.8缓冲溶液900mL为溶出介质,转速50rpm,经2、4、6、10、15、20、30、45、60min时,取溶液5mL,滤过,取续滤液用探索性研究建立的含量测定HPLC色谱条件进行溶出曲线研究。如图2所示,原研制剂在4种溶出介质中的溶出速率较快,在4种介质2min时的平均溶出量已达40%,B企业溶出行为与原研制剂较为相似,而A企业的制剂在4种溶出介质中15min前的溶出速率明显慢于其他两家企业的制剂。为考察溶出曲线差异是否由于原料药性质不同导致,本研究对各企业原料药的溶解度,固有溶出速率,被动转运的有效渗透性,LogP、LogD值、晶型、粒度等性质进行了测定与考察。结果显示,原料药不是造成溶出曲线差异的关键因素。通过崩解时限的考察,A企业样品崩解时限(5.6min)要远远高过原研制剂(0.9min)。推测A企业溶出速率慢于其他两家企业样品的原因为其采用湿法制粒引入较强粘合剂,造成片剂崩解较慢,考虑到药品的抗心绞痛功效,提示企业应调整工艺,提高药品的溶出速率。参照日本“橙皮书”条件,建立了溶出度HPLC检查方法,并用该方法测定了获得的88批次样品,结果显示两家企业样品溶出度均值分布较为分散,且溶出度结果相较国家药品标准测定结果均有不同程度降低,经与探索性研究建立的HPLC方法含量测定检测结果比对,溶出度低的主要原因为该批次样品含量偏低。
3.3 含量均匀度
取本品1片,置50mL量瓶中,加流动相适量,超声并振摇使尼可地尔溶解,用流动相稀释至刻度,摇匀,滤过,取续滤液作为供试品溶液,并用流动相配制相同浓度的对照品溶液,按建立的HPLC色谱条件(同“4.4含量测定”)检验88批样品。比较不同企业产品的含量均匀度A值与S值,与按现行国家标准检验结果不尽相同,A企业各批样品间的A值和S值均较大,显示其样品批间和批内差异均较大。而B企业各批样品间S值差异相对较大,A值差异相对较小,显示其样品批内差异较大。两企业样品均存在一定的工艺问题,建议相关企业加强生产工艺的研究,提高产品均一性。
4.4 含量测定
由于降解产物与主成分紫外最大吸收波长相近,故采用紫外E值法测定专属性较差。故建立HPLC方法,用十八烷基硅烷键合硅胶为填充剂(Inertsil ODS-3V C18,4.6×150mm,5µm),以甲醇-水(30∶70)为流动相,流速1.0mL·min-1,柱温30℃,以262nm作为检测波长,进样体积为20µL。精密称取约相当于尼可地尔10mg的样品,置100mL量瓶中,加流动相适量,超声并振摇使尼可地尔溶解,用流动相稀释至刻度,摇匀,滤过,取续滤液作为供试品溶液,并用流动相配制相同浓度的对照品溶液,共检验88批样品。两家企业样品测得的含量相较于紫外E值法测定的含量均有所降低,其中A企业降低更多,根据有关物质研究结果,这主要是由于A企业采用湿法制粒压片,制剂本身水分较高,同时其包装不足以隔离外部温湿度环境对样品的影响,使样品发生降解,进而导致了上述结果的产生。
4建议
通过研究发现尼可地尔片目前的相关药品质量标准及样品的处方、生产工艺和包装方面都存在若干问题。在质量标准方面本研究建议:①增订有关物质检查方法,制定合理的杂质限度;②修订含量测定和含量均匀度检查方法,将紫外E值法修订为专属性强的液相色谱法;③将溶出度检查方法由小杯法(《中国药典》2020年版附录0931第三法)-紫外E值法修订为桨法(《中国药典》2020年版附录0931第二法)-液相色谱法;④修订对检验员健康有风险的鉴别项,建立专属性强的液相色谱鉴别方法。在处方和生产工艺方面建议该品种仿制药生产企业按原研药调整处方,采用区分度高的溶出度控制方法进行质量控制,进行仿制药质量与疗效一致性评价。同时在处方中尽可能减少或避免使用羧甲基淀粉钠等具有引湿性的辅料,并尽量采用非湿法制粒工艺生产该品种,同时加强原辅料的水分控制及工艺过程中水分、温度等参数控制,以降低杂质产生量。在包装方面建议企业使用双铝箔板与自封式铝箔袋的包装,于铝箔袋中加入高效能干燥剂并在说明书及外包装中增加提醒患者在开封后保留干燥剂的说明。

来源:Internet