
嘉峪检测网 2025-09-09 20:42
导读:本文基于上述指南,结合行业实践案例,系统梳理清洁验证的核心框架与实施要点。
随着监管要求的不断严格与质量风险管理理念的普及,传统 “一次性验证” 模式已难以适应多产品共线、工艺持续优化的生产需求。2020 年 ISPE 发布《Cleaning Validation Lifecycle - Applications, Methods, and Controls》,首次系统提出清洁验证生命周期理念,将清洁活动贯穿于产品全生命周期;2025 年中国《清洁验证技术指南》进一步细化风险管理要求,明确全生命周期三阶段实施路径。本文基于上述指南,结合行业实践案例,系统梳理清洁验证的核心框架与实施要点。
清洁验证生命周期的理论基础与框架构建
1.1 清洁验证生命周期的溯源与核心原则
清洁验证生命周期理念源于质量源于设计(QbD)与全生命周期管理思想,其核心是将清洁验证从 “单一验证活动” 扩展为 “设计 - 确认 - 监控” 的持续改进过程。ISPE 2020 版指南与中国《清洁验证技术指南》共同确立了三大核心原则:
全生命周期覆盖原则:将清洁验证分为清洁工艺设计与开发(Stage 1)、清洁工艺验证(Stage 2)、持续清洁工艺确证(Stage 3)三个阶段,各阶段数据可反向反馈,实现工艺动态优化。
基于风险的决策原则:通过风险评估工具(如 FMEA、鱼骨图)识别清洁过程中的关键风险点(如难清洁设备、高毒性残留物),确定验证范围与深度,避免过度验证。
科学合规原则:优先采用基于健康的暴露限度(HBEL)等毒理学数据计算残留限度,替代传统 10ppm 或 1/1000 日治疗剂量法,提升限度设定的科学性。
1.2 传统清洁验证与生命周期方法的对比分析
传统清洁验证与生命周期方法在实施逻辑、关注点与灵活性上存在显著差异,具体对比见表。

清洁验证生命周期的三阶段实施路径
2.1 第一阶段:清洁工艺设计与开发(Stage 1)
本阶段是清洁验证的基础,核心任务是通过科学研究与风险评估,确定清洁工艺的关键参数与可接受标准,具体实施步骤如下:
残留物特性分析:识别待清洁污物的种类(产品活性成分、辅料、降解产物、清洁剂),评估其溶解性、吸附性、毒性等关键属性。例如,中药制剂需额外考虑原料的吸湿性与颜色残留,生物制品需关注蛋白质变性与微生物污染。
清洁工艺参数确定:基于 TACT(时间、作用、浓度、温度)原则设计清洁步骤,包括预处理(去除可见污物)、清洁剂洗涤(选择与设备材质相容的清洁剂)、冲洗(去除清洁剂残留)、干燥(控制微生物滋生条件)。例如,自动化清洁(CIP)需确定冲洗压力、温度与循环次数,手工清洁需明确擦拭力度与路径。
设备与取样方法设计:审核设备结构是否符合 “易清洁” 原则(如弧形角、无死角、抛光表面),通过风险评估确定取样点(优先选择难清洁区域,如反应釜底阀、搅拌杆)与取样方法(擦拭法或淋洗法)。同时,完成回收率研究,确保取样与分析方法的准确性(擦拭法回收率通常要求≥70%)。
残留限度计算:优先采用 HBEL 法计算最大允许残留总量(MAC),公式如下:\(MAC=\frac{PDE\times MBS}{LDD}\)其中,PDE 为每日允许暴露量(mg / 天),MBS 为下一产品最小批量(mg),LDD 为下一产品每日最大使用量(mg)。若毒理学数据不足,可采用 TTC(毒理学关注阈值)或传统 10ppm 法作为替代。
2.2 第二阶段:清洁工艺验证(Stage 2)
本阶段旨在通过实际运行证明清洁工艺的有效性与可重复性,如表:

2.3 第三阶段:持续清洁工艺确证(Stage 3)
本阶段是维持清洁验证状态的关键,通过日常监控与定期回顾,确保清洁工艺长期处于受控状态,具体措施如下:
日常监控:基于风险制定监控计划,包括常规取样检测(如每季度对难清洁部位进行擦拭取样)与工艺参数监控(如 CIP 系统的温度与压力报警记录)。监控数据需进行趋势分析,若出现接近警戒限的结果,需启动调查。
定期回顾:由跨职能团队(质量、生产、技术)每 1 - 2 年对清洁工艺进行全面回顾,内容包括:清洁工艺变更历史、偏差与纠正措施、微生物污染趋势、设备维护记录。若回顾发现工艺失控(如多次出现残留超标),需启动再验证。
变更控制:当发生设备改造、产品更换、清洁剂变更等情况时,需通过风险评估确定是否需要再验证。例如,引入更难清洁的产品时,需重新评估清洁工艺参数与残留限度,必要时补充验证批次。
清洁验证中的风险管理体系
3.1 风险评估工具与应用场景
清洁验证中常用的风险评估工具包括 FMEA、鱼骨图、HACCP(危害分析与关键控制点),其应用场景与核心目标如下:
FMEA:用于识别清洁过程中的潜在失效模式(如清洁剂浓度不足导致残留超标),通过评估严重性(S)、可能性(P)、可检测性(D)计算风险优先数(RPN = S×P×D),优先管控高 RPN 风险点。
鱼骨图:从 “人、机、料、法、环” 五个维度分析影响清洁效果的因素(如图 ),例如 “人员” 维度包括培训不足、操作不规范,“设备” 维度包括结构设计不合理、材质易吸附残留。
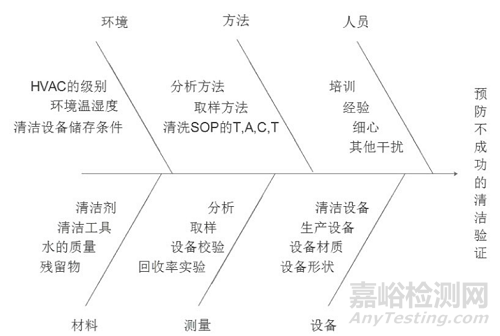
HACCP:识别清洁过程中的关键控制点(如清洁时间、取样位置),制定监控措施与纠偏行动,确保风险处于可控范围。例如,将 “清洁后设备保留时间(CHT)” 作为关键控制点,监控其是否超过预设限度(通常≤72 小时)。
3.2 关键环节的风险评估实践
产品与设备分组:通过风险评估将性质相似的产品(如溶解性、毒性相近)与设备(如结构、材质相同)分组,选取组内 “最差条件”(最难清洁产品、最难清洁设备)进行验证,减少验证工作量。例如,将 3 种水溶性、低毒性的原料药归为一组,选取其中吸附性最强的产品作为代表进行验证。
取样点风险评估:基于设备结构与清洗原理,优先选择 “高风险取样点”,包括:
难清洁区域:如管道焊接处、阀门密封面、搅拌轴密封
残留物易积聚区域:如反应釜底部、进料口拐角
微生物易滋生区域:如设备最低点(积水处)、复杂部件(如冻干机板层)
微生物风险管控:非无菌产品通常设定微生物负载限度(擦拭法≤50cfu/25cm²,淋洗法≤100cfu/mL),无菌产品需额外控制细菌内毒素。通过风险评估确定微生物监控频率,例如高湿度环境下需增加取样次数。
典型案例分析
4.1 原料药清洁验证案例
某企业生产 3 种原料药(A、B、C),共用 2 台相同玻璃反应釜(内表面积 8893cm²),清洁工艺为手工清洁(纯化水冲洗 + 丝光毛巾擦拭),具体验证步骤如下:
最差条件选择:通过溶解度(S)与吸附性(A)评分确定最难清洁产品。产品 C 溶解度最低(评分 4)、吸附性最高(评分 2),综合得分(S×A = 8)最高,被选为代表产品;设备选取反应釜 1 进行验证,反应釜 2 进行清洁确认。
残留限度计算:采用 HBEL 法计算产品 C 对产品 A 的 MAC 值为 17.79mg,擦拭取样限度为 5123.41μg/25cm²(警戒限 25.3μg/25cm²),淋洗取样限度为 364.5μg/mL(警戒限 1.80μg/mL)。
取样与检测:选取反应釜内壁、搅拌杆、底阀作为取样点,采用 HPLC 法检测化学残留,膜过滤法检测微生物。3 次验证批次结果均低于可接受限度,微生物负载≤20cfu/25cm²,验证通过。
4.2 无菌制剂清洁验证案例
某企业无菌制剂生产线生产 3 种注射剂(A、B、C),共用灌装加塞机(含陶瓷泵、灌装针头),清洁工艺为在线清洁(CIP),验证重点如下:
微生物风险控制:取样点优先选择灌装针头内壁(难清洁且直接接触药液),采用淋洗法取样,微生物限度设定为≤10cfu/100mL(注射用水淋洗),细菌内毒素≤0.25EU/mL。
清洁参数确认:CIP 工艺参数为:40 - 55℃纯化水冲洗 30min,0.1% 氢氧化钠溶液循环清洗 20min,注射用水冲洗至电导率≤2μS/cm。通过 3 次验证批次,确认该参数可有效去除残留(活性成分残留≤1μg/mL)。
持续监控计划:每批生产后检测灌装针头淋洗水的微生物与内毒素,每月对 CIP 系统参数进行回顾,确保工艺稳定性。
不同产品生命周期的共线清洁策略
5.1 临床研究阶段(I/II 期)
此阶段产品批量小、工艺未定型、毒理学数据不足,共线清洁策略以 “清洁确认” 为主:
设备选择:优先使用小型多功能设备或一次性耗材(如一次性反应釜),减少清洁难度;若使用商业化生产线,需对接触药液部件进行专用化处理(如更换密封圈)。
清洁确认要求:每批生产后进行清洁确认,采用非专属性方法(如 TOC、电导率)快速检测残留,若产品毒性较高(如细胞毒性药物),需补充专属性检测(如 HPLC 法)。
风险控制:制定严格的产品生产顺序(低毒性产品→高毒性产品),避免交叉污染;清洁后设备保留时间≤24 小时,防止微生物滋生。
5.2 商业化生产阶段(III 期 / 上市后)
此阶段产品工艺成熟、毒理学数据充分,共线清洁策略以 “清洁验证 + 持续监控” 为主:
共线评估:通过计算产品的 PDE 值评估共线可行性,若高毒性产品(如 PDE<10μg / 天)与低毒性产品共线,需采用专用清洁工艺或增加验证批次。
验证方案优化:采用 “分组法 + 最差条件法” 减少验证工作量,例如将 5 种水溶性产品归为一组,选取最难清洁产品进行 3 次验证,组内其他产品通过清洁确认即可。
持续改进:基于定期回顾数据优化清洁工艺,例如通过趋势分析发现某设备底阀残留易超标,可增加拆卸清洁步骤并补充验证。
5.3 研发与商业化产品共线
当研发产品(如拟上市品种)与商业化产品共线时,需采取 “分级管控” 策略:
生产计划隔离:研发产品与高风险商业化产品(如无菌制剂)错峰生产,间隔时间≥72 小时,避免交叉污染。
清洁验证要求:研发产品若为高毒性或难清洁品种,需单独开展清洁验证;若为低风险品种,可沿用商业化产品的清洁工艺,通过 1 次清洁确认证明适用性。
数据追溯:建立完整的共线生产与清洁记录,包括产品切换时间、清洁参数、检测结果,确保数据可追溯与监管合规。
结语
清洁验证生命周期管理是制药行业质量风险管理的重要组成部分,其核心价值在于通过 “设计 - 确认 - 监控” 的动态循环,实现清洁工艺的科学合规与持续改进。本文通过梳理三阶段实施路径、风险管理工具与共线策略,结合典型案例验证了该体系的实操性。

来源:Internet
关键词: 清洁验证