药物研发进程中,非临床药理毒理评价是评估候选化合物安全性的基石,为首次人体试验(FIH)提供至关重要的科学依据。该评价体系需遵循《药物非临床研究质量管理规范》(GLP),其核心在于系统揭示药物的吸收、分布、代谢、排泄(ADME)特性及潜在毒性风险。不同类型药物的研究策略虽有差异,但核心评价框架保持一致。
临床前药理毒理评价有哪些内容?
•《药物非临床研究质量管理规范》(国家食品药品监督管理总局令第34号) 指出非临床安全性评价研究,指为评价药物安全性,在实验室条件下用实验系统进行的试验,包括:安全药理学试验、单次给药毒性试验、重复给药毒性试验、生殖毒性试验、遗传毒性试验、致癌性试验、局部毒性试验、免疫原性试验、依赖性试验、毒代动力学试验以及与评价药物安全性有关的其他试验。
单次给药毒性试验(急性毒性)
核心目标: 明确单次或24小时内多次给药后产生的急性毒性反应特征及其可逆性,识别主要毒性靶器官。
核心价值: 为重复给药毒性试验的剂量设计、临床起始剂量选择及急性中毒风险评估提供关键参考。
实验设计: 需采用两种相关动物种属(特定情况如部分中药可用一种)、两种性别、健康成年动物。剂量设计方法多样(如最大耐受量法、固定剂量法、上下法等)。观察期通常不少于14天,密切监测临床症状、死亡、体重变化,终末进行大体解剖和组织病理学检查。
关键产出: 最大无毒性反应剂量(MFD)、最大耐受剂量(MTD)、剂量-反应关系、靶器官初步判断。
重复给药毒性试验(长期毒性)
核心目标: 全面描述动物长期重复暴露于受试物后的毒性反应谱,确定毒性靶器官、剂量依赖性和时间进程。
核心价值: 预测人体潜在不良反应、确定未观察到有害作用水平(NOAEL)、推导FIH的起始安全剂量范围。
实验设计: 要求两种相关种属、两种性别动物。设置低(通常接近或略高于预期有效暴露量)、中、高(应能诱导明显毒性反应)剂量组及溶剂对照组。给药频率通常每日一次,试验周期需覆盖并支持拟开展临床研究的期限。监测指标全面,包括行为学、摄食量、体重、眼科、体温、心电图(ECG)、临床病理学(血液学、血生化、尿液分析)及详尽的组织病理学检查。
关键产出: 明确NOAEL、毒性靶器官、可逆性评估、安全范围界定。需结合毒代动力学(TK)数据进行综合评价。
核心目标: 检测受试物是否引起DNA损伤及其固定,评估潜在的致突变和致癌风险。
适用范围: 中药、天然药物、化学药物基本要求。
标准策略: 通常采用以下组合之一:
组合一: 细菌回复突变试验(Ames试验) + 体外哺乳动物细胞遗传损伤试验(如染色体畸变试验、微核试验) + 一项体内遗传毒性试验(通常为啮齿类骨髓微核试验)。
组合二: Ames试验 + 两项使用不同组织的体内遗传毒性试验(如骨髓微核试验 + 肝脏彗星试验)。
关键产出: 综合判断受试物是否具有遗传毒性潜力。
核心目标: 评估药物对亲代生殖功能(配子发生、交配行为、受孕、妊娠、分娩、哺乳)及子代发育(胚胎-胎儿发育、出生后生长发育直至性成熟)的潜在不良影响。
核心价值: 是评估药物生殖发育安全性的关键环节,对临床用药人群(尤其是育龄人群)的风险管理至关重要。
试验分期: 通常按生育力与早期胚胎发育(I段)、胚胎-胎仔发育(II段)、围产期发育(III段)分阶段进行,覆盖完整生殖发育周期。
关键考量: 需根据药物特点、临床适应症和拟用人群确定试验策略和时机(参见ICH S5(R3)等指南)。常伴随TK研究(母体、胚胎/胎仔、乳汁暴露)。
安全药理学
核心目标: 识别药物在治疗剂量及以上范围内,对生理功能(特别是生命体征相关系统)产生的非预期药效学作用(不良反应)。
适用范围: 普遍适用于所有药物(含生物制品)。
核心组合: 重点关注中枢神经系统(FOB)、心血管系统(清醒动物血压、心率、ECG,体外hERG)和呼吸系统(频率、潮气量等)的功能评价。
实验设计: 通常在清醒动物上进行,单次给药,剂量需涵盖并超出治疗剂量范围以达到充分暴露。根据核心组合结果或化合物特性,可能需进行追加或补充试验(如对泌尿、胃肠系统的影响)。
关键产出: 评估药物对重要生命功能系统的潜在风险。
核心目标: 识别受试物在动物中的潜在致癌性,并评估其对人体的相关风险。
启动依据: 通常基于长期用药(≥6个月或频繁间歇性长期使用)、特定担忧因素(如阳性遗传毒性结果、明确免疫抑制或激素活性、特定患者人群、高全身暴露、结构警示、光致癌潜力等)进行评估。
实验系统: 首选大鼠,通常需第二种啮齿类(小鼠)。特殊情况下可使用替代模型。
关键产出: 评估肿瘤发生率、类型、潜伏期、剂量-反应关系,并与人体药代动力学特征进行关联性分析,判断人体风险。
局部毒性试验
核心目标: 评估非口服给药途径(如注射、皮肤、粘膜、眼、吸入等)对用药局部组织产生的刺激性、腐蚀性、过敏性反应,以及对全身产生的过敏性、溶血性等不良反应。
关键内容: 局部刺激性/过敏性试验、全身过敏性试验、溶血性试验。
关键产出: 明确给药途径相关的局部和全身安全性风险。
免疫原性试验
核心目标: 评估治疗性蛋白、多肽及其衍生物(如抗体偶联药物ADC)诱发抗药抗体(ADA)的能力及其潜在后果(如中和活性、过敏反应、影响药效/清除)。
研究重点: ADA的检测与表征(发生率、滴度、持续时间、中和能力)。
检测策略: 采用层级化的分析方法(筛选、确证、滴度测定、中和活性分析),常用方法包括桥联法、直接法等。
关键产出: 评估免疫原性风险及其对临床安全性和有效性的潜在影响。
依赖性试验
核心目标: 评估药物产生精神依赖(渴求)和躯体依赖(戒断症状)的潜力,警示潜在滥用倾向。适用范围: 主要针对作用于中枢神经系统(CNS)的药物。
评估策略: 早期评估(结构分析、靶点预测、PK特性)结合依赖性行为学试验(药物辨别、自身给药、戒断评价),常用大鼠模型。
实验设计: 通常设阳性/阴性对照,在最高预期人体治疗剂量Cmax附近设置剂量,在药物达峰时间(Tmax)附近观察行为学变化。关键产出: 判断药物的依赖性潜力等级。
毒代动力学
核心目标: 在毒性试验中同步测定受试物在动物体内的全身暴露水平(AUC, Cmax等)及其持续时间,建立暴露量与毒性效应的关系。
核心价值: 解释毒性反应、支持种属选择合理性、优化给药方案、量化靶器官暴露、桥接动物与人体安全性(为FIH剂量选择提供依据)、指导临床安全监测。
实施要点: 伴随主要毒性试验(尤其是重复给药毒性试验)进行,关注关键参数(AUC0-t, Cmax, C(time)),确保分析方法可靠。
关键产出: 暴露量-毒性关系分析,支持非临床到临床的安全性转化。
支持IND申报所需的试验
支持IND申报的关键非临床研究概览
为成功提交IND申请,需整合并提供以下关键非临床研究数据:
药理学: 体内外主要药效学(作用机制验证、概念验证POC)。
分析学: 经过验证的药物制剂分析方法和生物样品分析方法。
药代动力学: 全面的体内ADME研究及体外代谢/相互作用研究。
毒理学(安全性评价核心):
安全药理学(核心组合:CNS、心血管、呼吸系统)。
一般毒性试验:剂量范围探索试验(DRF)及符合GLP规范的单次和重复给药毒性试验(试验期限不短于拟开展的临床研究给药期)。
遗传毒性试验(标准核心组合)。根据药物特性和临床计划,可能需要的其他试验(生殖毒性早期试验、局部耐受性、免疫原性、依赖性潜力初步评估等)。
制剂安全性: 根据给药途径和制剂类型,完成相应的局部耐受性/刺激性/过敏性等试验。
总结
非临床药理毒理评价是一个系统、严谨且高度法规化的过程。通过精心设计和执行上述关键研究,旨在全面阐明候选药物的安全性特征,识别潜在风险点(毒性靶器官、安全范围、特殊毒性如遗传、生殖、致癌、依赖风险等),并利用毒代动力学数据建立动物与人体暴露关联,为首次人体临床试验的安全启动奠定坚实的科学基础,最终保障受试者安全。各项研究的具体实施需严格遵循国内外相关技术指导原则的要求。
支持IND申报的非临床研究主要清单
|
研究类别 |
核心目的/内容 |
关键产出/目标 |
|
体内外药理学 |
|
|
|
分析方法开发与验证 |
药物制剂分析、生物样本(如血浆)中药物及代谢物定量分析方法 |
|
|
药代动力学(PK) |
体内ADME过程、体外代谢(酶抑制/诱导)及转运体研究 |
|
|
安全药理学(核心组合) |
|
|
|
单次给药毒性试验(急性) |
|
确定MTD/MFD,识别急性靶器官,支持起始剂量设计 |
|
重复给药毒性试验(长期) |
重复给药的毒性特征(符合GLP,期限≥临床拟用期限) |
确定NOAEL/靶器官/可逆性,推导临床安全起始剂量及范围 |
|
遗传毒性试验(核心组合) |
评估致突变和染色体损伤潜力(通常:2个体外 + 1个体内试验) |
|
|
制剂安全性试验 |
根据给药途径(如注射、外用、眼用等)评估局部耐受性 |
确保特定剂型和给药方式下的局部与全身安全性(如刺激性等) |
|
(酌情增加) |
|
|
|
生殖毒性试验(早期阶段) |
|
|
|
免疫原性试验(生物制品) |
|
|
|
依赖性潜力评估 |
|
|
附表:试验清单
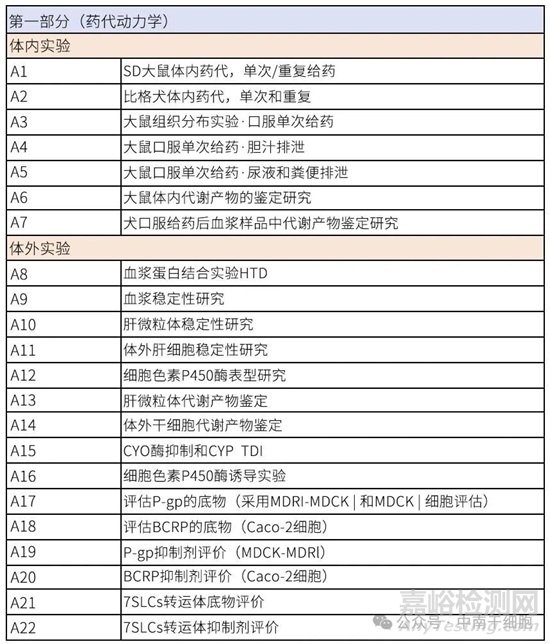


来源:Internet
关键词:
IND申报药理毒理所需试验清单
