概述:对于非无菌化学药品及原辅料,微生物限度是反映产品安全性和质量可控性的重要指标之一。
一、非无菌原辅料微生物限度研究
原辅料微生物控制策略的建立:
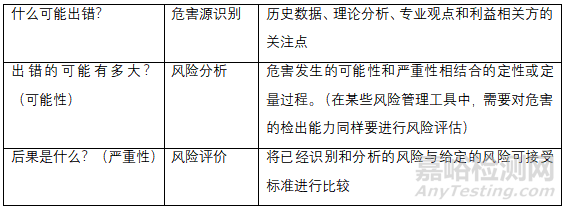
原辅料微生物限度研究应基于风险评估,风险评估方法可以参考风险评估方法表和公认的风险管理工具表或适用合理的方法。
风险评估方法表
首先企业需要对自己的产品做到心里有数,然后针对以上的三个问题进行评估,而风险评价要综合以上三个基本问题且有详实的数据作为强有力的支撑,这决定了评估结果的质量。
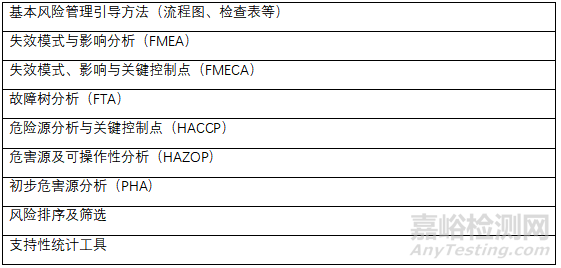
公认的风险管理工具
上述工具可适用于DS和DP质量等相关的特定领域。质量风险管理方法和支持性统计工具可以组合使用,有助于质量风险管理原则的灵活应用。
● 风险评估需综合考虑原辅料的性质、物料性质(包括起始物料、溶剂、试剂、催化剂等)、生产工艺、车间环境、设备清洁状态、人员素质、最差生产条件、历史数据及趋势等因素,参考ICH Q6A(原料药和辅料的微生物限度检查)制定微生物控制策略。
● 首先判断微生物能否在原辅料中生长或存活,若有足够的科学支持性数据表明微生物既不能生长也不能存活,结合风险评估结果可不考虑对微生物限度进行检测。反之,应进行研究和检查。
● 需要说明的是,某些情况下原辅料不支持微生物的生长和繁殖,但仍需要对微生物限度进行研究和控制。例如,一些水分活度低的原辅料不适宜微生物的生长和繁殖,但如果原辅料的初始生物负载较高,某些耐受性较强的微生物仍可能存活并引入到终产品中。
二、非无菌化学药品微生物限度研究
微生物控制策略的建立:
* 当制剂中含有抑菌剂或制剂本身具有抗菌效力时,应进行如下研究:
● 含有抑菌剂,应制定抑菌剂浓度可接受范围,通过抑菌效力试验来证明低于或等于拟定的抑菌剂最低浓度时的有效性;
● 或提供科学依据证明制剂本身具有抗菌效力。需制定合理的微生物限度可接受标准,并对微生物限度进行逐批检测。
1)若每批微生物限度检测结果均符合可接受标准,可进行定期抽样检测,抽样频率通过风险评估结果确定,或提供科学依据证明可不考虑进行常规微生物检测。
2)若定期抽样检测样品出现微生物检测结果大于可接受标准或微生物增长趋势明显的情况,应及时进行原因调查并将控制策略调整为逐批检测。
3)若微生物限度检测结果大于可接受标准或微生物增长趋势明显时,则需将微生物限度订入注册标准并逐批检测。
4)需要说明的是,对于部分非无菌化学药品如口服溶液剂、栓剂等中国药典中明确需进行微生物限度检查的剂型,应结合中国药典相关要求将微生物限度订入注册标准逐批检测。
* 如制剂中不含抑菌剂或制剂本身不具有抗菌效力,应判断制剂是否为固体制剂(如,口服固体制剂)。
● 如制剂属于固体制剂,且具有足够的科学依据证明其具有抑制微生物生长的特性,可不考虑进行微生物限度检查。
● 如制剂不属于固体制剂,或虽属于固体制剂但没有足够的科学依据证明其具有抑制微生物生长的特性,可参照以上1)和2)两步执行。
检测方法的选择:
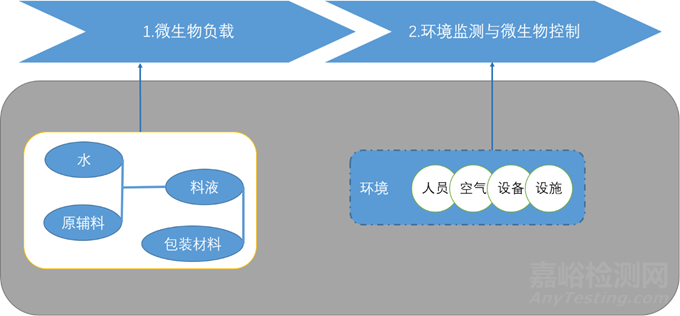
微生物负载控制策略:
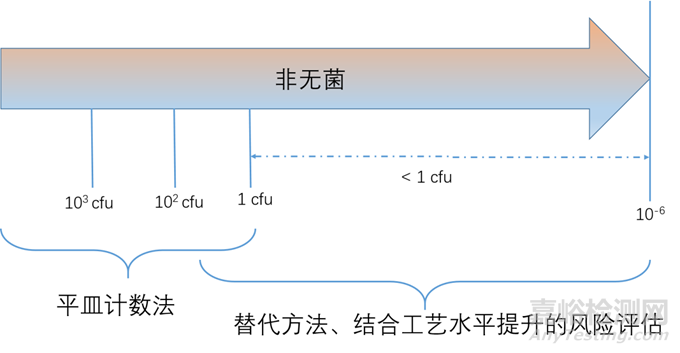
在原辅料生产过程的质量控制或终产品放行过程中,其方法与药典方法中相比,某些参数可能存在差异,但二者应有明确的相关性;当药典方法难以满足质量控制要求时,可根据风险评估结果,采用经验的替代方法进行质量控制。当替代方法应用于终产品放行时,其应优于或等同于药典方法。
限度的制定:
● 综合考虑中国药典相关要求、原辅料来源、性质、生产工艺条件、给药途径及微生物污染对患者的潜在危险、目标患者人群等因素。
● 对于特殊品种(如,小规格的吸入制剂)可在对上述因素评估的基础上,考虑以最小包装单位规定限度标准,并提供相应的风险评估资料。对药品成份中含有动物内脏提取物、未经提取的动植物来源成分及矿物质,或与上述产品共线生产时,还应对沙门菌进行检测。
● 境外上市药品申请进口时,应结合产品具体剂型,原则上不低于中国药典的要求;或根据风险评估结果,结合中国药典要求,制定合理的控制策略。
非无菌药品微生物限度标准可以参考《中国药典》2020版通则(1107);微生物负载和微生物限度之间区别和联系:微生物限度是根据患者的给药途径,而微生物负载则是基于生产工艺控制需要;两者之间的联系都是采用微生物计数法。
生产过程中的微生物控制:
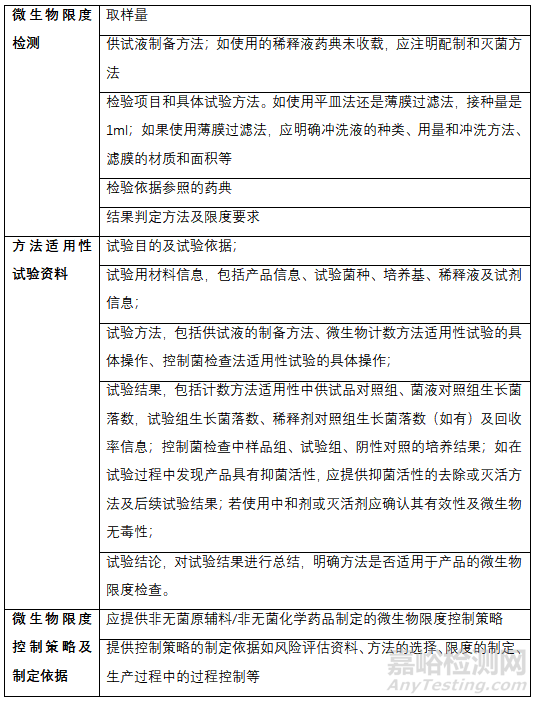
三、微生物限度申报资料要求
申报资料内容

来源:注册圈
关键词:
微生物限度
化学药品