
嘉峪检测网 2024-05-08 07:59
导读:什么是DHF,什么是DMR,似乎还有很多同行未曾接触,该概念来源于21 CFR P820。
什么是DHF,什么是DMR,似乎还有很多同行未曾接触,该概念来源于21 CFR P820。
GB/T 42061中 4.2.3 有关于医疗器械文档的要求。GMP中第二十四条提到技术文件和记录,这里的技术文件是指产品的DMR,记录则应是包含两类的记录,一类是产品开发过程中的DHF记录,一类是生产过程中的DHR记录。
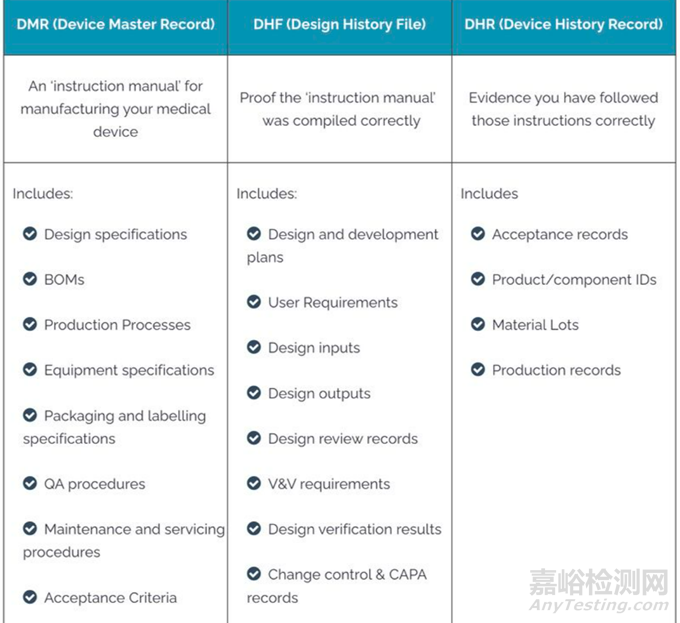
医疗器械的DHF文件(Design History File,设计历史记录)在医疗器械开发和制造过程中似乎比DMR文件(Device Master Record,产品主文档)更为重要。
是因为DHF在整个器械生命周期中的关键作用,从产品概念到市场投放,都需要依赖于DHF文件的完整性和准确性。
怎么说?
在医疗器械领域中,每一件产品从设计到生产,再到最终的市场投放,都必须遵循严格的规范和程序,以确保其安全性和有效性。
在众多相关文件中,设计历史文件(DHF)和产品主文档(DMR)是两类核心文件,它们分别承载着产品开发历史的详细记录和产品生产的标准要求。虽然DMR在生产过程中发挥着基础的作用,但DHF因其涵盖的广泛性和深入的历史追溯功能,在整个产品生命周期管理中显得尤为重要。
DHF的定义与重要性:
DHF是记录医疗器械从设计到市场投放全过程的文件,它不仅记录了设计和开发的每一步,还包括了验证和验证活动的详细记录。这些信息集合体是监管机构审查的重要依据,同时也是企业内部确保产品符合原始设计规范和法规要求的关键文件。DHF提供了一条清晰的证据链,证明产品是按照适当的设计控制程序开发的。
DHF通常包括以下几个核心组成部分:
(1)设计输入与输出:这些记录确保产品设计满足预定的使用需求和预期的应用场景。设计输入是基于市场需求、用户反馈、前期技术评估和监管标准制定的。而设计输出则是实际的产品规格,应直接反映设计输入的要求。
(2)设计审核记录:在产品开发的多个阶段,进行设计审核以确保设计过程符合预定的质量标准,同时满足用户和监管需求。
(3)设计验证与验证活动:验证活动确认设计输出符合设计输入的要求,而验证则确保产品能在实际使用中安全有效地执行预定功能。
(4)风险管理文件:产品设计和开发过程中的风险评估和控制措施记录,包括风险分析、风险评估以及风险缓解策略。
(5)变更控制记录:任何设计变更都需要经过严格的审查和记录,确保变更不会对产品的性能和安全产生负面影响。
为何DHF比DMR更重要:
(1)审查与合规性:监管机构在审查医疗器械上市申请时,DHF提供了一个全面的产品开发历史视图。通过评审DHF中的信息,审查员可以详细了解产品从概念到最终实现的全过程,评估产品是否符合所有相关的医疗设备标准和法规。
(2)质量控制与产品完整性:DHF作为记录产品开发生命周期的文档,是质量控制的关键。它详细记录了产品设计和开发过程中的每一个决策和变更,对于追踪产品问题、实施纠正措施及进行持续改进至关重要。
(3)历史数据的保存与追溯:在产品面临法律诉讼或召回时,DHF提供了不可争辩的历史数据支持,这些数据对于解决争议、应对监管审查及指导未来的产品改进都极为重要。
总结:尽管DMR在保证生产过程符合设计规格和保障产品一致性方面发挥着重要作用,但DHF在整个产品的生命周期管理中起着更加全面和决定性的作用。它不仅关系到产品的合规性、效能和安全性,还直接影响到企业的声誉、产品的市场表现以及最终的商业成功。因此,DHF的作用和重要性在很大程度上超过了DMR,是医疗器械企业管理和监管过程中不可或缺的核心文件。

来源:医械研发
关键词: 医疗器械文件