
嘉峪检测网 2024-05-31 11:39
导读:本文主要结合作者几年来的CTD资料撰写的工作经验,依据ICH Q3A中对新原料药的杂质定义并结合案例对化学合成原料药杂质谱分析的一般原则和检查方法的研究。
摘要:杂质作为药品研发的一项关键质量属性,对药物纯度有着重要的影响,制定质量控制策略,建立合理的检查方法是研发过程中研究的重要内容。
新原料药:先前尚未在任何成员国或地区注册的具有治疗作用的活性成分(也称为新分子或新化学实体)。它可以是某种已获批准的药物的一种复合物、简单的酯或盐。
杂质谱:对存在于某一新原料药中的已鉴定或未鉴定杂质的数量及含量。
原料药的杂质谱分析对应CTD格式申报资料中的 M3.2.S.3.2杂质(不考率安慰剂的情况下)。
本文主要结合作者几年来的CTD资料撰写的工作经验,依据ICH Q3A中对新原料药的杂质定义并结合案例对化学合成原料药杂质谱分析的一般原则和检查方法的研究。
杂质杂质分类及来源:
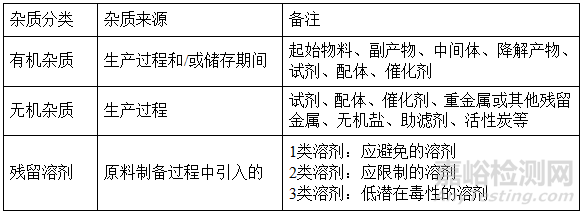
根据ICH Q3C指导原则,对原料药制备过程中引入的所有溶残的测定方法进行研究,并建立相应的限度,各残留溶剂的控制限度与ICH Q3C(R5)“杂质,残留溶剂指导原则”推荐限度一致。
溶剂残留的杂质限度:

杂质图谱分析案例-经验分享
使用1H-NMR 对内标进行测定
如下图所示;
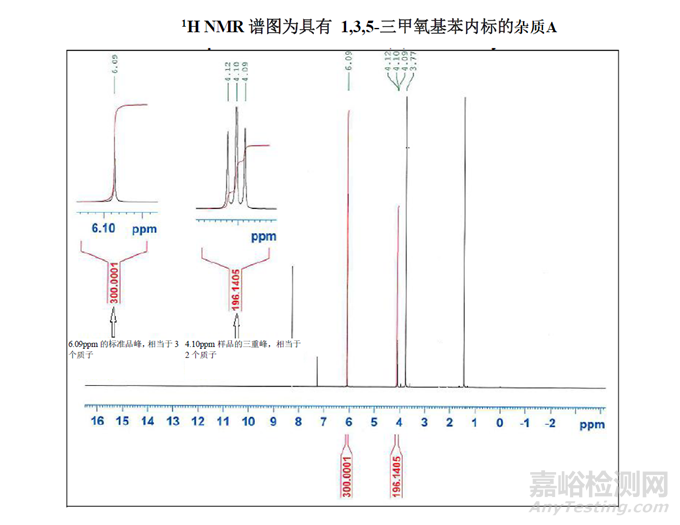
该项目中选择1,3,5-三甲氧基苯作为内标,因为考虑到它的峰不会对杂质A样品中存在的任何峰造成干扰,并在CDCl3中制备样品,使两种材料都以等摩尔量的形式存在。

5秒是所有峰返回正值所需的最短时间。根据计算,其中T大约等于tnull/ln(2),新原料中杂质(假设该杂质为A)分析的时间T将为14秒。松弛延迟应≥ (7/3)*T,相当于大约17秒。建议增加5-10秒作为安全余量。因此,该方法中将延迟设置为30秒。
样品溶液在t=0 时提交,测定结果为99.8%。20小时后重新提交相同的样品,检测结果为100.8%。由于差异≤ 2.0%,因此认为样品溶液可稳定长达20小时。
该方法的运行结果证明,该方法适用于1H-NMR 测定杂质A。
HPLC法:
1.首先确定色谱条件,包括色谱柱的选择(在合成工艺中,分析方法纯化使用C18的硅胶柱比较多,仅针对于此杂质,色谱柱的选择应结合杂质本身的理化特性,进行筛选,对于相对稳定的物质多选用C18硅胶色谱柱),流动相的确定(根据分子本身的极性、溶解度等性质进行确定,实验室内多用水和乙腈进行分析纯化),稀释剂的选择(稀释剂尽可能的选择与流动相溶剂相同,为了降低对设备的损耗或因溶剂的选择而致使产品分析不纯等情况)
2.溶液制备
3.图谱分析
经验分享:
以下通过三种分析方法对某一新原料药中关于杂质B纯度进行分析
方法一:等度洗脱
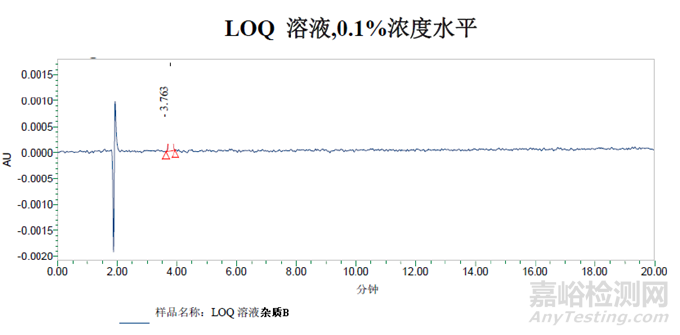
0.1%浓度水平色谱峰的信噪比为61。
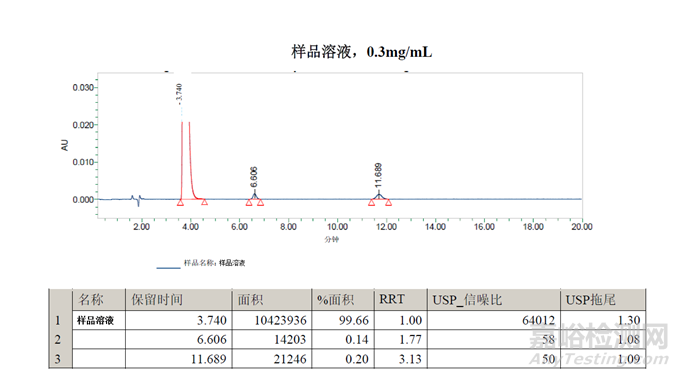
样品溶液在6.6分钟处检测到的峰可能是二氯-杂质B,但需要通过保留标记进行确认。在11.7分钟处检测到的峰是未知峰。
方法二:梯度
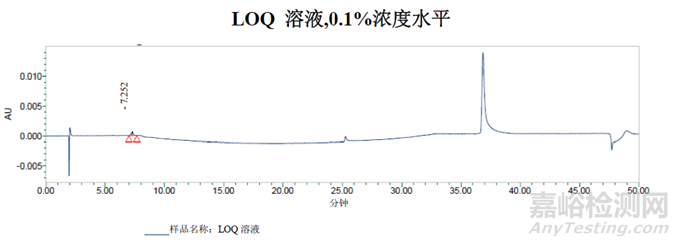
0.1%浓度水平色谱峰的信噪比为61。
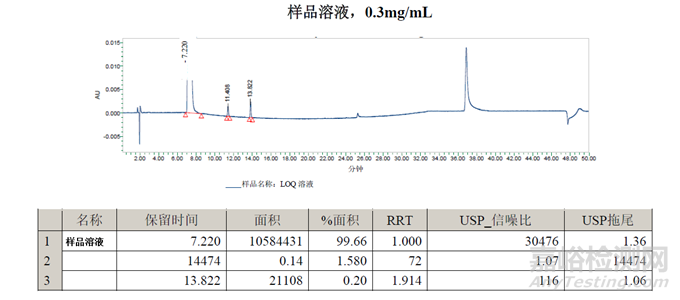
样品溶液的面积%数据与方法一一致。在11.4分钟处检测到的峰可能是二氯-杂质B,但需要通过保留标记进行确认。在13.8分钟处检测到的峰是未知峰。

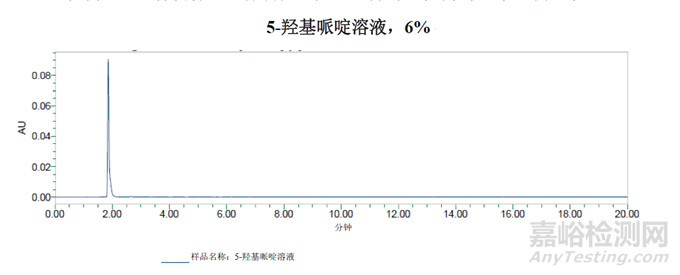
从5-羟基哌啶(6%)的稀溶液中获得的峰似乎并未保留,而是随进样峰被洗脱为切分峰。
通过对杂质图谱的分析得出的结论是:用于杂质B纯度测定的方法二并不适用于测定5-羟基哌啶。稀释剂需要更高的含水量,以防止早期洗脱组分的峰切分。梯度也需要从含水量更高的条件开始,为了从进样峰中分离出5-羟基嘧啶峰。
方法三:自行进行分析方法开发并经过验证
对方法二进行修改,通过更换流动相A,并同时改变稀释剂的配比,增加稀释剂的极性,从而防止峰切分。方法二中的其他条件保持不变。
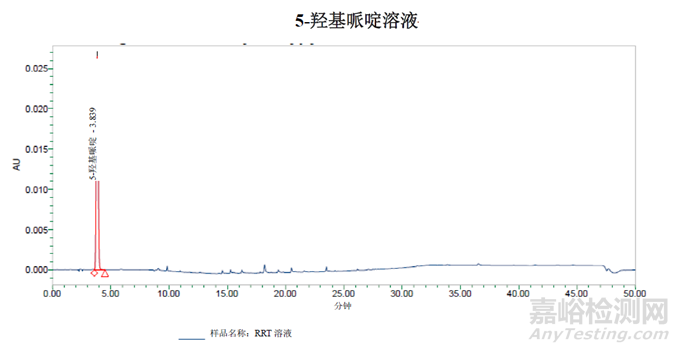
使用含水量更高的条件,从进样峰中分离出5-羟基哌啶峰未观察到切分峰。
结论:但是通过对方法三做出的修改似乎足以确定杂质B的纯度。
pH值测定法:
样品将以溶剂(溶剂的选取应是可以与水进行互溶溶剂)和水层的双相混合物形式提供。将样品转移至分液漏斗中,使各相分离。
将底部(水层)收集到适当大小的烧杯中,并测量pH值和温度。读数应记录在20-25℃之间。
KF库伦滴定法:
首先应进行溶液的配制,其次将配制好的样品放置设备中进行分析(应多次进样进行水分测定),根据公式计算水的含量,并确保重复检测结果符合以下要求,< 0.10%不限,0.10-1.0%绝对误差在0.2%以内,> 1.0%绝对误差在0.5%以内。
根据下列公式用仪器计算水含量:
含水量 = R-(B+(D ⅹ T))
式中:
水(R) = 消耗量(mC)/10.72(其中10.72 = mC,相当于1μg水)
D = 偏移(μg/min)
T = 滴定时间(分钟)
注册申请中应提供书面文件,证明分析方法是经过验证并适用于杂质的检测和定量(可以参见ICH Q2A及Q2B分析方法论证指导原则项下)。技术因素(如生产能力与质控方法)可作为部分依据来论证或选择其他的杂质限度。如果研发中所采用的分析方法和准备上市产品的分析方法不同,在申报资料中应予以讨论。

来源:药研
关键词: 新原料药杂质谱