
嘉峪检测网 2024-09-27 08:13
导读:建立高效液相色谱法在不同检测波长处同时测定阿咖酚散/胶囊中3种主要杂质成分水杨酸、对氨基酚、对氯苯乙酰胺的含量。
摘 要: 建立高效液相色谱法在不同检测波长处同时测定阿咖酚散/胶囊中3种主要杂质成分水杨酸、对氨基酚、对氯苯乙酰胺的含量。采用Ultimate LP-C18 (150 mm×4.6 mm, 5 µm)色谱柱,进行多波长分析。流动相为磷酸盐缓冲液(0.01 mol/L磷酸二氢钾溶液,用磷酸调节pH值至2.6±0.1)-甲醇(7∶3),水杨酸、对氨基酚、对氯苯乙酰胺检测波长分别为241、263、254 nm,流量为0.9 mL/min,柱温为30 ℃,进样体积为20 µL。3种杂质的质量浓度在各自范围内与色谱峰面积的线性关系良好,相关系数均为0.999 8,定量限为0.03~0.14 µg/mL。样品加标平均回收率为97.7%~102.3%,测定结果的相对标准偏差均小于3.0%(n=6)。该法可用于阿咖酚散/胶囊中3种主要杂质成分的含量测定。
关键词: 多波长; 高效液相色谱法; 阿咖酚散/胶囊; 杂质检测
阿咖酚散/胶囊作为常用的感冒药,内容物主要为每粒含阿司匹林230 mg、对乙酰氨基酚126 mg、咖啡因30 mg和辅料适量的复方制剂。因其价格便宜在市场上流通广泛,临床上主要用于解热镇痛[1]。由于生产阿咖酚散/胶囊的厂家较多,简单的包装在生产、运输、储存中可能产生降解杂质。目前实施的质量标准有国家食品药品监督管理局国家药品标准WS-10001-(HD-0941)-2002和YBH 01232010,其中,检查项只对阿司匹林的降解产物游离水杨酸进行杂质控制,而对主成分对乙酰氨基酚中可能存在的降解产物对氨基酚、对氯苯乙酰胺未建立限度检查[2‒3]。由于对氨基酚、对氯苯乙酰胺等杂质成分具有遗传毒性、肾毒性等[4‒5],对人体健康存在一定隐患,且阿咖酚散/胶囊的现行标准对杂质水杨酸的检查方法不同、限度(散剂为2.0%,胶囊剂为3.0%)控制不同。
笔者通过文献研究[6‒14]发现阿咖酚散/胶囊的质量控制不够全面准确、杂质检查方法不一,故建立了多波长高效液相色谱法[15]同时检测阿咖酚散/胶囊中3种主要杂质成分的含量,能够快速准确地进行定量检测,为阿咖酚散/胶囊的杂质控制及质量标准提升提供依据。
1、 实验部分
1.1 主要仪器和试剂
高效液相色谱仪:LC-20AT型,日本岛津公司。
紫外分光光度计:UV-2700型,日本岛津公司。
超声波清洗器:KQ-500E型,昆山市超声仪器有限公司。
电子天平:XS205DU型,感量为0.01 mg,瑞士梅特勒-托利多公司。
超低温冷冻储存箱:DW-FL450型,中科美菱低温科技有限公司。
甲醇、乙腈:均为色谱纯,美国默克密理博公司。
磷酸:优级纯,国药集团化学试剂(北京)有限公司。
磷酸二氢钾:分析纯,国药集团化学试剂(北京)有限公司。
阿咖酚散/胶囊:共11批,样品信息及生产厂家见表1。
表1 样品信息及生产厂家
Tab. 1 Sample information and manufacturer

水杨酸、对氨基酚、对氯苯乙酰胺标准品:纯度(质量分数)分别为99.8%、98.2%、100.0%,批号分别为100106-202106、100802-202306、100850-202304,中国食品药品检定研究院。
实验用水:超纯水。
1.2 色谱条件
色谱柱:Ultimate LP-C18 柱(150 mm×4.6 mm, 5 µm,美国Welch公司);柱温:30 ℃;进样体积:20 µL;流动相:A相为磷酸盐缓冲液(取0.01 mol/L磷酸二氢钾溶液,用磷酸调解pH值至2.6±0.1),B相为甲醇,等度洗脱,流量为0.9 mL/min;液相洗脱方法见表2。
表2 液相洗脱方法
Tab. 2 Elution method of HPLC
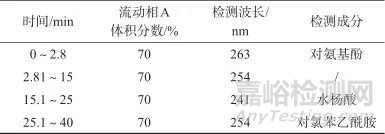
1.3 溶液制备
1.3.1 混合对照品溶液
在避光条件下分别精密称取水杨酸标准品54.06 mg、对氨基酚标准品8.02 mg、对氯苯乙酰胺标准品4.05 mg,置于同一20 mL棕色量瓶中,加入甲醇适量,振摇使其溶解,并定容至标线,作为混合标准储备液。
精密量取混合标准储备液5 mL,置于同一100 mL棕色容量瓶中,加甲醇稀释并定容至标线,摇匀,作为混合标准溶液。
1.3.2 系列混合标准工作溶液
精密量取混合标准储备液适量,用甲醇稀释成系列混合标准工作溶液,其中水杨酸的质量浓度分别为270.30、135.15、67.58、33.79、16.89、3.38、0.68 µg/mL,对氨基酚的质量浓度分别为40.10、20.05、10.02、5.01、2.51、0.50、0.10 µg/mL,对氯苯乙酰胺质量浓度分别为20.25、10.12、5.06、2.53、1.26、0.25、0.05 µg/mL。
1.3.3 样品溶液
在避光条件下,取《中华人民共和国药典》2020年版四部“通则0115散剂”中“装量差异”项下内容物(10袋共含阿司匹林2 300 mg、对乙酰氨基酚1 260 mg、咖啡因300 mg和辅料适量的复方制剂,批号为230101),混合均匀,精密称取适量(约相当于对乙酰氨基酚100 mg),置于10 mL棕色容量瓶中,加入甲醇适量,超声溶解后,定容至标线,摇匀滤过,作为样品溶液。
1.3.4 加标样品溶液
在避光条件下,取《中华人民共和国药典》2020年版四部“通则0115散剂”中“装量差异”项下内容物,混合均匀,精密称取适量(约相当于对乙酰氨基酚100 mg),置于10 mL棕色容量瓶中(平行取样18份),分别精密加入混合对照品储备液2.0、2.5、3 mL各6份,再加入甲醇,定容至标线,摇匀,滤过。
1.3.5 空白溶液
以甲醇作为空白溶液。所有溶液均在-30°C超低温冷冻储存箱下储存。
1.4 实验方法
取表1中不同生产厂家的11批样品按1.3.3方法制备样品溶液,每批各2份,再按照1.2色谱条件进样测定,利用标准工作曲线外标法计算3种杂质成分的含量,水杨酸含量按阿司匹林标示量的百分比计算,对氨基酚及对氯苯乙酰胺含量按对乙酰氨基酚标示量的百分比计算,得到3种杂质的测得量(质量分数)见表3。
表3 样品杂质含量(质量分数)测定结果
Tab. 3 Determination results of impurity content (mass fraction) in the sample ( % )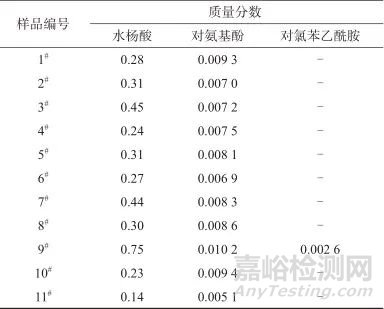
注:“-”表示未检测出。
2、 结果与讨论
2.1 测定波长及溶剂选择
经紫外分光光度计全波长扫描,发现水杨酸、对氨基酚、对氯苯乙酰胺在240~270 nm波长处有最大吸收,经液相色谱进样比较系统适用性参数后选择测定波长分别为241、263、254 nm,各待测成分峰形、响应值和分离度均较好。对比国家食品药品监督管理局国家药品标准WS-10001-(HD-0941)-2002和YBH 01232010中单波长等度检测,结果发现采用多波长法切换检测条件下,各成分系统适用性参数更佳,因此选择多波长法。
分别考察了甲醇、乙醇、水作为溶剂对测定结果的影响。结果发现甲醇作为溶剂时,样品在1 min内超声能够完全溶解,而乙醇、水作为溶剂时样品超声较难溶解。溶解时间长可能会增加阿司匹林水解,导致杂质水杨酸含量增大,因此采用甲醇作为溶剂,结果发现样品峰形等各项色谱参数均符合测定要求。
2.2 流动相及柱温选择
考察磷酸盐缓冲液在不同pH值下(pH值分别为2.0、2.4、2.6、3.0)与甲醇配制的流动相,发现pH<2.6时,对氨基酚与对乙酰氨基酚色谱峰重叠,pH=3.0时,水杨酸的稳定性不佳,因此选择磷酸盐缓冲液pH=2.6。考察不同比例的流动相及0.8、0.9、1.0 mL/min的进样流量,结果发现磷酸盐缓冲液-甲醇(7∶3)、流量0.9 mL/min的条件下,各待测成分分离度最佳。通过比较不同柱温下(25、30、35 ℃)混合对照品溶液中各待测物峰面积响应及理论塔板数,结果见表4。
表4 不同柱温下各成分的峰面积及理论塔板数
Tab. 4 Sample impurity content determination results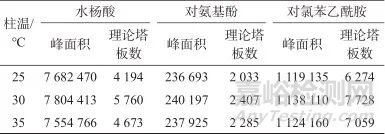
由表4可知,柱温为30 ℃时各待测成分峰面积及理论塔板数最大,因此选择柱温为30 ℃。
2.3 专属性
取空白溶液、混合对照品溶液、加标样品溶液、样品溶液按照1.2色谱方法进样,色谱图分别见图1~图4。由图4可知,各成分专属性强,其他原辅料成分不影响各待测物质的含量测定。对氨基酚、水杨酸、对氯苯乙酰胺先后出峰,理论塔板数均大于2 000,分离度均大于2.0。

图1 空白溶液色谱图
Fig. 1 Chromatogram of blank control solution
t/min
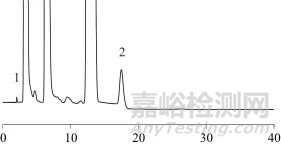
图4 样品溶液色谱图
Fig. 4 Chromatogram of sample solution
1—对氨基酚;2—水杨酸
t/min
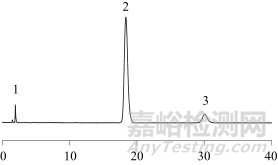
1—对氨基酚;2—水杨酸;3—对乙酰氨基酚图2 混合对照品溶液色谱图
Fig. 2 Chromatogram of mixed standard solution
t/min

1—对氨基酚;2—水杨酸;3—对乙酰氨基酚图3 加标样品溶液色谱图
Fig. 3 Chromatogram of spiked sample solution
t/min
2.4 线性关系与定量限
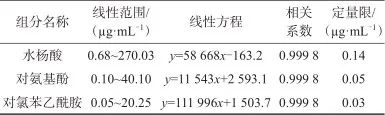
按照1.2色谱方法测定系列混合标准工作溶液。以质量浓度(x)为横坐标,色谱峰面积(y)为纵坐标绘制标准工作曲线,利用加权最小二乘法进行回归运算。以信噪比为10作为各成分的定量限。3种杂质成分的线性范围、线性方程、相关系数及定量限见表5。由表5可知,水杨酸、对氨基酚、对氯苯乙酰胺的质量浓度在各自范围内与色谱峰面积具有良好的线性关系,相关系数均为0.999 8,定量限分别为0.14、0.05、0.03 µg/mL,表明该方法具有较高的灵敏度,适于定量。
表5 线性回归方程及定量限结果
Tab. 5 Linear regression equation and quantitative limit results
2.5 稳定性试验
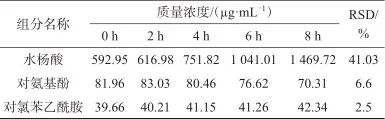
取1.3.3中含混合标准储备液2.0 mL的加标样品溶液,于室温条件下分别避光放置0、2、4、6、8 h,在1.2色谱条件下进样测定,记录色谱峰面积并带入线性方程计算各成分的质量浓度,试验结果见表6。由表6可知,样品溶液中杂质水杨酸随阿司匹林降解含量逐渐增加;对氨基酚在4 h内较为稳定,超过4 h后含量逐渐降低;对氯苯乙酰胺在8 h内稳定,测定结果的相对标准偏差小于3.0%,因此样品测定时应临用现配,配制时间控制在2 h以内并立刻进样。
表6 稳定性试验结果
Tab. 6 Stability test results
2.6 样品加标回收率与精密度试验
在避光条件下,选取已知浓度的加标样品溶液,按照1.2色谱方法进样并测定,记录色谱峰面积,计算加标回收率及测定结果的相对标准偏差,试验结果见表7。由表7可知,水杨酸加标平均回收率为99.9%~102.3%,对氨基酚的加标平均回收率为97.7%~100.8%,对氯苯乙酰胺的加标平均回收率为98.1%~100.7%,测定结果的相对标准偏差均小于3.0%,表明该方法具有较高的准确度和良好的精密度,能够满足实际检测的需要。
表7 样品加标回收试验结果
Tab. 7 Sample addition recovery test results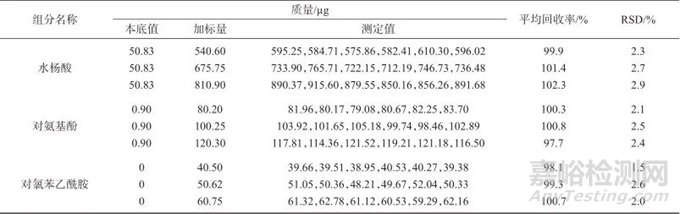
3、 结语
以紫外光谱扫描的最大吸收波长为参照,采用多波长高效液相色谱法同时检测阿咖酚散/胶囊中可能存在危害人体健康的3种主要杂质。该方法灵敏度高、稳定性好,对阿咖酚散/胶囊杂质控制方面的不足加以补充,可为阿咖酚散/胶囊质量控制标准提升提供技术参考。
参考文献:
1 李万平,万莉,张华,等.阿咖酚散中游离水杨酸检查方法的改进[J].中国药师, 2017, 20(8):1 494.
LI Wanping,WAN Li,ZHANG Hua,et al. Improvement of detection method of free salicylic acid in Agafol powder[J]. Chinese Pharmacist,2017, 20(8):1 494.
2 廖燕萍. 阿咖酚散微生物计数方法的建立与研究 [J]. 海峡药学, 2018, 30(11):60.
LIAO Yanping. Establishment and study of microbiological counting method of Acafol powder[J]. Straits pharmacy,2018, 30(11):60.
3 赵淑晶,高永坚,区淑蕴,等. HPLC法测定对乙酰氨基酚原料药有关物质及遗传毒性杂质[J]. 广东药科大学学报,2022,38(3):13.
ZHAO Shujing,GAO Yongjian,QU Shuyun,et al. Determination of paracetamol related substances and genotoxic impurities by HPLC[J]. Journal of Guangdong Pharmaceutical University,2022,38(3):13.
4 KHANDAVILLI UBR, KESHAVARZ L, SKOŘEPOVÁ E,et al. Organic salts of pharmaceutical impurity p-aminophenol[J]. Molecules, 2020, 25(8):1 910.
5 曾金亮,周雅丽,庞发根,等.复方氨酚溴敏胶囊中对氨基酚的检查[J].广州化工, 2023, 51 (11):122.
ZENG Jinliang,ZHOU Yali,PANG Fagen,et al. Examination of aminophenol in compound aminophenol bromin capsules[J].Guangzhou Chemical,2023, 51(11):1 22.
6 胡海侠,高燕,宏伟.高效液相色谱多波长同时测定阿咖酚散中对乙酰氨基酚、咖啡因、阿司匹林、水杨酸[J].化学分析计量,2021,30(5):37.
HU Haixia,GAO Yan,HONG Wei,et al. Determination of acetaminophen, caffeine, aspirin and salicylic acid in Acafol powder by high performance liquid chromatography with multiple wavelengths[J]. Chemical Analysis Meterage,2021,30(5):37.
7 江坤,陈颜清,蔡锦雄,等. HPLC法测定氨酚伪麻那敏分散片中的对氨基酚[J].中国药品标准, 2022, 23 (1):24.
JIANG Kun,CHEN Yanqing,CAI Jinxiong,et al. Determination of paraminophenol in paraminophenol and pseudomanamine dispersive tablets by HPLC[J]. Chinese Drug Standard,2022, 23 (1):24.
8 王燕桓,刘国瑞,傅承光,等. HPLC法测定阿咖酚散中3种有效成分及相关杂质含量的研究[J].药物分析杂志,2006,26(5):692.
WANG Yanheng,LIU Guorui,FU Chengguang,et al. Determination of 3 active components and related impurities in Agafol powder by HPLC[J]. Journal of Pharmaceutical Analysis,2006,26(5):692.
9 肖江,王辉,帅海涛,等.高效液相色谱法检测对乙酰氨基酚片中有关物质[J].中国药业,2022,31(21):55.
XIAO Jiang,WANG Hui,SHUAI Haitao,et al. Determination of related substances in acetaminophen tablets by high performance liquid chromatography[J]. Chinese Pharmaceutical Industry,2022,31(21):55.
10 刘晶晶,梁智渊,陈承贵,等. HPLC法测定氨咖黄敏胶囊的有关物质[J]. 中国药师,2021,24(9):1 769.
LIU Jingjing,LIANG Zhiyuan,CHEN Chenggui,et al. Determination of related substances in ankahuangmin capsules by HPLC[J]. Chinese Pharmacist,2021,24(9):1 769.
11 王英瑛,李俊,杨玉琴. HPLC法测定复方对乙酰氨基酚片中对氨基酚和对氯苯乙酰胺的含量[J].中国药师,2018,21(7):1 303.
WANG Yingying,LI Jun,YANG Yuqin,et al. Determination of p-aminophenol and p-chloroacetamide in compound paracetamol tablets by HPLC[J]. Chinese Pharmacist,2018,21(7):1 303.
12 程侠,吴毅彦,郝桂明,等.氨酚咖匹林片中对氨基酚和对氯苯乙酰胺的检测[J].天津药学,2021,33(2):9.
CHEN Xia,WU Yiyan,HAO Guiming,et al. Determination of para-aminophenol and p-chlorophenol acetamide in para-aminophenol and capilline tablets[J]. Tianjin Pharmacy,2021,33(2):9.
13 肖羽,陈安丽,尹玮璐,等.高效液相非接触电导法测定阿咖酚散3种有效成分的含量[J].今日药学,2016,26(1):33.
XIAO Yu,CHEN Anli,YIN Weilu,et al. Determination of three active components in Acafol powder by high performance liquid phase non-contact conductance method[J]. Pharmacy Todayt,2016,26(1):33.
14 国家药典委员会.中华人民共和国药典(2020年版)[M]. 4部.北京:中国医药科技出版社,2020.
National Pharmacopoeia Commission. Pharmacopoeia of the People's Republic of China(2020 Edition)[M]. Part 4. Beijing:China Medical Science and Technology Press,2020.
15 姜军华,陈建兰,许妍,等. HPLC多波长切换法同时测定小儿咽扁颗粒中11种成分的含量[J].中国药事,2023,37(2):199.
JIANG Junhua,CHEN Jianlan,XU Yan,et al. Simultaneous determination of 11 components in Xiaoer Yanbian granules by HPLC multi-wavelength switching method[J].Chinese Pharmaceutical Affairs,2023, 37 (2):199.
引用本文: 曹梦欣,臧晴晴 . 多波长高效液相色谱法同时测定阿咖酚散/胶囊中3种杂质成分[J]. 化学分析计量,2024,33(8):79. (CAO Mengxin, ZANG Qingqing. Simultaneous determination of three impurity components in Akafen powder/capsule by multi-wavelength high performance liquid chromatography[J]. Chemical Analysis and Meterage, 2024, 33(8): 79.)

来源:化学分析计量
关键词: 胶囊杂质检测