
嘉峪检测网 2024-08-15 09:07
导读:分析中国加入国际人用药品注册技术协调会(ICH)后,国内企业仿制药中美双报注册的现状及其注册要求,以期为国内企业提供参考。
摘要
目的 分析中国加入国际人用药品注册技术协调会(ICH)后,国内企业仿制药中美双报注册的现状及其注册要求,以期为国内企业提供参考。方法 比较分析中国和美国化学仿制药注册申报路径、通用技术文档(commontechnical document, CTD)、药学研究和生物等效性研究等方面的具体要求,总结我国企业中美双报的优势和劣势,强调中美双报过程中需要关注的重点。结果 2017 年以来,国内企业在美国的仿制药申请(abbreviated new drugapplication, ANDA)获批数量显著增加;中美仿制药注册在CTD申报的要求、药学研究资料、生物等效性试验的设计等方面有差异也有共性,国内企业在注册申报时需对这些差异予以重视,深入理解两国的技术要求。结论 从研发立项、质量标准、参比制剂选择、生物等效性试验设计等方面分别提出了建议,以期为我国企业中美双报提供参考,加快药品在两国上市速度,提升国内企业的国际竞争力。
关键词:中美双报;化学仿制药;比较分析
我国加入国际人用药品注册技术协调会(ICH)为实现药品注册技术要求与国际规则协调统一创造了良好的政策环境。在进行药物研发及目标上市市场确定时,国内企业申报策略越来越倾向于中美双报。仿制药是与原研药具有相同的活性成分、剂型、给药途径和治疗作用的药品[1]。仿制药中美双报是指用同一套项目研究资料,同时或分阶段在中国与美国进行仿制药注册申报[2]。我国的参比制剂包括原研药品以及国际公认具有参比制剂地位的制剂,当中含有美国橙皮书所列清单药品,这为仿制药中美双报提供了可能。加快以中美双报为代表的制药国际化,把握中美双报注册关键点,对于节约企业成本和时间、加快药品在两国上市速度、提高企业国际竞争力和推动医药产业高质量发展具有重要意义。
一、我国企业在美国申报仿制药现状
根据药智网数据显示,2014—2022年国内企业获 FDA 批 准 的 仿 制 药 申 请(abbreviated new drugapplication, ANDA)数量大幅增加,自 2018 年起维持在 100个以上,2022年获批数量为 2014年的 5.41倍,这表明国内企业国际药品注册申报技术能力不断提升,见图1。从各企业获批数量来看,中美双报已经培育出 一批具有全球竞争力的企业,2014-2022 年ANDA 获批数量排名靠前的企业有浙江华海药业、复星医药、南通联亚、南京健友、人福美国、宜昌人福、以岭药业、齐鲁制药、石药欧意和杭州海正,这些企业ANDA获批数量总计为618个,占总获批数量的76.3%。
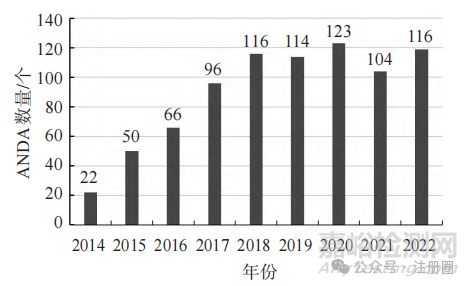
▲图1-中国企业在美国获批的ANDA数量
二、化学仿制药中美注册对比分析
中美两国仿制药研发都以质量源于设计(quality by design, QbD)理念为有效研发工具,仿制药的药学研究目的都是确保产品与参比制剂质量与疗效一致,但因两国特定监管体系和政策方向的影响,在注册申报和技术要求方面也存在一些不同[3]。本 文从注册申报途径 、通用技术文档(common technical document, CTD)、药学研究和生物等效性研究等方面,对化学仿制药在中国与美国注册申报进行对比分 析,以寻求中美双报的可行性。
2.1 仿制药申报的途径和方法
我国《化学药品注册分类及申报资料要求》明确了仿制药包括化药3类、化药4类和化药5.2类[4];申请人应当参照相关技术指导原则选择合适的参比制剂,进行生物等效性(bioequivalence, BE)备案并开展相关试验[1]。申请人递交仿制药上市申请至国家药品审评中心,该中心首先进行形式审查,形式审查通过后发放受理通知书、注册检验通知书及缴费通知书等,之后进入技术审评阶段,待通过技术审评及国家药品监督管理局要求进行的注册检验核查等其他相关程序后,企业将获得该药品的批准文号,成为药品上市许可持有人。
根据美国《联邦食品、药品和化妆品法案》505部分,适用于仿制药上市许可的申请为 505(j)即ANDA,包含能证明仿制药与参比制剂具有相同活性成分、剂型、给药途径和治疗作用等相关的资料[5]。ANDA 申请人需根据《联邦法规》第21章节的具体要求准备 ANDA申报资料,该章节下的320部分内容为生物利用度(bioavailability, BA)和生物等效性要求。具体来看,320 部分的 320.21BE 递交要求、320.22 体内 BE 豁免要求、314.94ANDA 递交内容及格式和 314.70 批准后药品变更内容规定均直接适用于 ANDA 申报[6]。FDA 下属仿制药办公室(office of generic drugs, OGD)负责对申请人递交的ANDA申报资料进行审评,ANDA审评周期一般为10个月,美国ANDA审评流程见表1。
▲表1-ANDA审评流程
2.2 注册代理机构
我国仿制药的申请人应当为能够承担相应法律责任的企业或者药品研制机构等,这表明注册代理机构可由直接参与药品研发的机构或者企业承担。美国仿制药申请要求境外申请人指定在美国境内居住或者有商业办公地址的公司或者个人作为美国代理,美国代理负责与FDA进行沟通。对于进行仿制药中美双报的企业而言,选择合适的美国代理并建立有效的沟通机制是实现快速且成功上市的关键因素。
2.3 直接接触药品的包装材料和容器(以下简称原辅包)
中美两国都通过法律法规、规范性文件构建了原辅包注册管理制度框架,原辅包的注册申报资料均需要按照CTD格式整理并提交申报资料[7]。
我国除了仿制境内已上市药品所用化学原料药可单独审评,其余情况都需要与制剂关联审评。原料药生产商在原辅包平台进行登记,制剂注册申请时与已登记原辅包进行关联。美国进行仿制药申请之前,需要完成药品主文件(drug master file,DMF)备案,DMF通过FDA完整性评估后才可以被仿制药申请引用与制剂进行关联审评,DMF未通过完整性评估,ANDA存在被拒收风险。中美双报自制原料药的质量需分别符合《中国药典》和《美国药典》标准,并在中国与美国进行注册登记。
2.4 CTD文档
ICH M4 为人用药品注册 CTD 文档,注册申报资料分为 5 个模块[8],主要内容见表 2。eCTD 是将符合CTD规范的药品申报资料电子化,能够实现电子申报资料提交、接收、验证、受理、审评和电子存档全过程操作,使药品全生命周期管理和文档储存更加简洁明了,提高了审评机构审评效率,促进药品全球同步研发[9]。国家药品监督管理局于2020年发布《化学药品注册分类及申报资料要求》,明确了所有药物的临床试验、药品上市注册及化学原料药申请均需按照 ICH M4 的 CTD 格式进行申报[4]:模块一为区域性文件,因不同国家地区而异;ANDA不要求非临床研究,所以一般不包含模块四[10-11];中国、美国仿制药申报模块二与模块五区别在于我国目前实行电子资料申报,而美国实行eCTD申报,两国CTD模块二与模块五资料要求见表3。
▲表2-仿制药申请CTD主要内容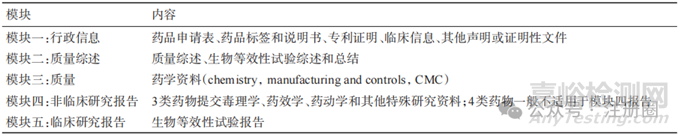
▲表3-中美两国注册申报资料模块二与模块五对比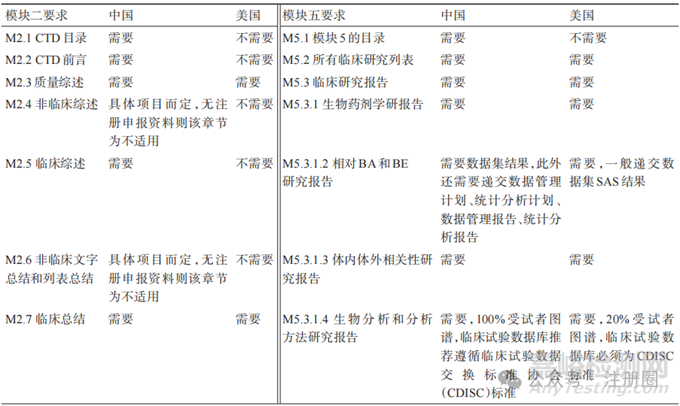
2.5 药学资料(CMC)
CMC主要包括产品以及参比制剂前期研发、处方工艺研究、质量研究、稳定性研究等,是药品申报资料非常重要的组成部分。关于 CMC中美两国都强调仿制药的质量必须符合国家药典和相关法规的要求,在一些细则上仍存在差异,CMC 典型差异见表4。
▲表4-中美两国注册申报资料CMC对比
2.6 生物等效性试验(BE)方案设计
《化学药物制剂人体生物利用度和生物等效性研究技术指导原则》指出生物等效性研究方法优先考虑程度最高的是药动学研究方法[12]。按照药动学为评价方法的中美 BE 研究设计的差异见表5。《药品注册管理办法》中规定,药品注册申请人应当在药品审评中心网站完成备案后方可开展 BE 研究;FDA则对BE无备案要求。关于BE给药状态,FDA针对缓释胶囊类药品需进行空腹撒布性药物生物等效性研究,空腹试验时,将药物洒在苹果酱上给吞咽胶囊有困难的患者吞服,例如卡马西平缓释胶囊、盐酸维拉帕米缓释胶囊[13]。关于清洗期,FDA规定给药间隔不短于 5 个消除半衰期,我国规定不短于7个消除半衰期。中美 BE 试验结果评价标准一致,为仿制药中美双报提供了较大可行性。
▲表5-中美两国注册申报资料BE试验方案对比
三、国内企业仿制药中美双报的优势和劣势
3.1 优势
第一,节约成本和时间,基于同一套研究资料,在中国和美国同时或分阶段进行注册申报,可以节省重复研发和申报的资源。
第二,加快药品在两国市场的上市速度。
第三,提升国际的药品注册申报能力。
第四,增强企业的品牌影响力以及国际市场竞争力。
3.2 劣势
第一,中美两国在药品注册的法规、申报资料内容、注册代理机构、生物等效性试验方案设计等方面存在差异,药品注册申报资料需要满足两国的不同要求,增加注册过程复杂性。
第二,资源分散,在中美两国市场同时进行注册申报会影响研发和市场策略的聚焦。
第三,仿制药在中国、美国注册的申报途径、审评周期存在差异,企业需要根据两国设定具体的注册申报时间规划,以免影响产品整体上市速度。
四、中美双报策略建议
4.1 选题立项
以满足临床需求为宗旨进行流行病学调查,确保产品在中美两国的目标市场上有一定的临床需求;了解各目标市场上已有类似适应证品种的近年销售数据、不同产品处方以及规格的调查分析;对产品专利情况进行全面文献检索,有助于企业前瞻研判。
4.2 质量标准
企业在制定质量标准时,应综合考虑国家药品监督管理局和 FDA 发布的指导原则、《中国药典》、《美国药典》以及参比制剂的质量要求,从而确保所拟定的质量标准既符合中方的要求,也能满足国际化的发展趋势。在药品研发过程中,企业应持续关注药典标准和其他参考资料的最新动态,确保研发活动的调整和优化。随着生产工艺的进步和完善,企业还应不断提升质量标准,以保证药品的质量和疗效,满足患者的需求和期望。
4.3 参比制剂
化学仿制药研发,参比制剂的选择至关重要。2022年度药品审评报告指出,仿制药注册申请研发立项方面存在的主要问题为化学仿制药开发时参比制剂选择不当[14]。中美化学仿制药申报必须选择在国家药品监督管理局发布的化学仿制药参比制剂目录内且在美国橙皮书列出名单的药品作为参比制剂。仿制药中美双报选择参比制剂时,还应关注药品规格、药品处方信息,了解药品安全性、有效性和药品适应证能否满足两国的临床需求等。若原研药品在美国撤市或者未获得参比制剂地位则不能作为参比制剂;对于未在国家药品监督管理局发布的参比制剂目录内的药品,需要在国家药品监督管理局药品审评中心进行参比制剂遴选申报,建议企业等待所选产品列入国家药品监督管理局发布的参比制剂目录之后再进行下一步研究,避免参比制剂经审评后未被纳入参比制剂目录而造成经济损失。
4.4 原辅包登记
当企业进行仿制药中美双报时,应密切关注原辅包材料在两国市场上的登记情况、DMF申报状态和药典的更新情况,确保分别符合《中国药典》和《美国药典》要求。我国属于原料药生产和出口大国[15],大部分原料药获得了美国 DMF 备案[16],因此中美两国在原料药方面的差异比较小。在药用辅料与药包材方面,我国关联审评制度的核心要素内涵,如登记主体完整性审查、技术审评等与美国DMF登记制度基本一致,且在辅料与药包材变更管理申报上,中美都以药品上市许可人为责任主体评估辅料变更的影响程度并决定是否申报管理[17]。但是两国在辅料登记范围与登记状态内涵上也存在差异,企业在仿制药中美双报时应予以重视[18]。
4.5 生物等效性试验(BE)
2022年度药品审评报告还指出,仿制药注册申请有效性方面存在的主要问题为 BE方案无法证明其和参比制剂的疗效一致性。企业要以临床需求为导向,对化学仿制药中美双报 BE 设计异同加以关注。例如,2021 年药品审评中心发布《醋酸阿比特龙片生物等效性研究技术指导原则》指出本品与食物同时服用时,阿比特龙全身暴露量升高,需进行空腹人体生物等效性研究[19];FDA 仿制药生物等效性的具体指南数据库关于醋酸阿比特龙片生物等效性草案建议申请人开展空腹以及餐后2个状态下的生物等效性研究[20]。因此,仿制药BE设计要同时满足两国试验设计要求,建议申请人根据 BE 方案单独保存数据库,如空腹和餐后 2 个不同试验设计成为单独的试验方案,构建单独的英文数据库。
4.6 eCTD软件和人员
仿制药申请过程中,美国及其他40多个国家均采用eCTD申报,凸显了eCTD在全球统一标准方面具有先进优势,未来各类药品注册申请、医疗器械等 eCTD 申报也是大势所趋。2021 年,国家药品监督管理局正式发布《国家药监局关于实施药品电子通用技术文档申报的公告》,规定化药药品注册分类 1类、5.1类以及治疗用生物制品 1类和预防用生物制品1类可以按照eCTD进行申报[21],暂未要求仿制药上市申请实行eCTD申报。2023年1月1日起,我国已全面实行电子申报;2023年12月11日,药品审评中心发布关于更新《申报资料电子光盘技术要求》等文件的通知,对电子申报的要求进一步细化;目前,我国处于电子申报资料递交和 eCTD 递交并存的阶段。仿制药中美双报需要相关专业人员熟练操作 eCTD 信息化系统。当前,我国制药行业相关人员以及药品监管机构审评人员熟悉eCTD资料编制和出版的专业人才较少。一方面,药品审评中心要加大对 eCTD 相关培训与宣讲力度、优化注册申报系统、发布常见问答,帮助企业更好地理解我国以及国外 eCTD 的技术要求和申报流程[22];另一方面,企业需要提早布局,组建专业eCTD注册申报团队,增强国际注册申报竞争力,提升药品的注册申报成功率。
4.7 加强沟通交流
我国现行的《药物研发与技术审评沟通交流管理办法》已适用于创新药、改良型新药、生物类似药、复杂仿制药以及一致性评价品种等研发过程和注册申请中的沟通交流[23];FDA 有专门针对仿制药的正式沟通交流会议的指南[24],指南明确了沟通交流会议的具体技术要求,FDA期望通过加强申请人与审评部门的沟通交流以缩短审评周期使得药品尽快获批上市。因此,仿制药中美双报时应积极申请沟通交流会与当局沟通,提高仿制药审批通过率。
参考文献
[1] 国务院办公厅 . 关于开展仿制药质量和疗效一致性评价的意见[EB/OL]. [2023-12-10]. https://www.gov.cn/xinwen/2016-03/05/content_5049517.htm.
[2] 祁鸽, 刘文轩, 李亦兵 . 中美双报重点考量因素及申报策略建议[J]. 中南药学, 2022, 20(1):226-229.
[3] 周玉琦. 从行政法的视角探究中美药品注册审批制度[J]. 长沙民政职业技术学院学报, 2013, 20(3):51-55.
[4] 国家药品监督管理局 . 总局关于发布化学药品注册分类改革工作方案的公告(2016 年第 51 号)[EB/OL]. [2023-12-10].https://www. nmpa. gov. cn/xxgk/ggtg/ypggtg/ypqtggtg/20160309151801706.html.
[5] U. S. Food and Drug Administration. FD&C Act Chapter V:Drugs and Devices[EB/OL]. [2023-12-10]. https://www. fda.gov/regulatory-information/federal-food-drug-and-cosmetic-actfdc-act/fdc-act-chapter-v-drugs-and-devices.
[6] U.S. Food and Drug Administration.Code of federal regulations title 21-food and drugs[EB/OL]. [2023-12-10]. https://www.fda.gov/medical-devices/medical-device-databases/code-federal regulations-title-21-food-and-drugs.
[7] 王佳, 袁利佳, 张宁 . 中美原料药、药用辅料和药包材的注册管理对比研究[J]. 中国药物评价, 2021, 38(6):488-492.
[8] 国家药品监督管理局 . 关于发布《M4:人用药物注册申请通用技术文档(CTD)》模块一文件及 CTD 中文版的通告(2019年第 17 号)[EB/OL]. [2023-12-10]. https://www.nmpa.gov.cn/xxgk/ggtg/ypggtg/ypqtggtg/20190417174001488.html.
[9] 江惠, 王丽洁, 郭晓迪 . 中国化学仿制药全球注册申报的现状和趋势[J]. 中国医药工业杂志, 2020, 51(12):1594-1601.
[10] 李岩 . 关于完善我国仿制药品注册管理体制的研究[D]. 沈阳:沈阳药科大学, 2009.
[11] DA ANDA Submissions. Content and format of abbreviated new drug applications [A/OL]. [2023-12-10]. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/anda-submissions-content-and-format-abbreviated-new-drugapplications.
[12] 国家药品监督管理局药品审评中心 . 化学药物制剂人体生物利用度和生物等效性研究技术指导原则[EB/OL]. [2023-12-10]. https://www. cde. org. cn/zdyz/domesticinfopage? zdy‐zIdCODE =90be26c277291c580525 d2a50c45ae6b.
[13] 刘倩, 南楠, 李涛, 等 . FDA《特定药物的生物等效性指导原则》内源性物质药物生物等效性指导原则介绍[J]. 中国新药杂志, 2018, 27(5):509-514.
[14] 国家药品监督管理局药品审评中心. 2022年度药品审评报告[EB/OL]. [2023-12-10]. https://www.cde.org.cn/main/news/viewInfoCommon/849b5a642142fc00738aff200077 db11.
[15] 贺刘莹, 杨锐, 王晓锋, 等 . 高端药物制剂用特殊功能辅料的研究进展[J]. 中国药学杂志, 2023, 58(3):197-204.
[16] 李寅, 张帆 . 全球仿制药、原料药行业发展趋势[J]. 科学观察, 2012, 7(6):1-10.
[17] 袁利佳, 汪小燕, 王佳, 等 . 关联审评政策下药用辅料与药包材变更管理的思考[J]. 中国药事, 2022, 36(2):121-127.
[18] 吕旭峰, 袁利佳, 王佳, 等 . 我国药用辅料关联审评制度改革进展及思考[J]. 中国药事, 2024, 38(2):123-129.
[19] 国家药品监督管理局药品审评中心 . 醋酸阿比特龙片生物等 效 性 研 究 技 术 指 导 原 则[EB/OL]. [2023-12-10]. https://www. cde. org. cn/zdyz/domesticinfopage? zdyzIdCODE =d3ae2c04614 d6bed443 d33bc5a28e874.
[20] U.S.Food and Drug Administration. Product-specific guidancesfor generic drug development[EB/OL]. [2023-12-10]. https://www.fda.gov/drugs/guidances-drugs/product-specific-guidances-generic drug-development.
[21] 国家药品监督管理局 . 关于实施药品电子通用技术文档申报的公告(2021 年第 119 号)[EB/OL]. [2023-12-10]. https://www. nmpa. gov. cn/xxgk/ggtg/ypggtg/ypqtggtg/20210930111641179.html.
[22] 陈华, 邢花. 我国正式实施eCTD后药品生产企业的注册申报应对策略[J]. 中国新药杂志, 2023, 32(12):1177-1184.
[23] 国家药品监督管理局药品审评中心 . 关于发布《药物研发与技术审评沟通交流管理办法》的通告(2020 年第 48 号)[EB/OL]. [2023-12-10]. https://www.cde.org.cn/main/news/viewInfoCommon/b823ed10 d547b1427a6906c6739fdf89.
[24] 崔晶, 董旻, 吕旭峰, 等 . 美国 FDA 复杂仿制药注册沟通交流制度研究及启示[J]. 中国药学杂志, 2022, 57(24):2135-2138.

来源:广东药科大学学报
关键词: 化学仿制药