
嘉峪检测网 2024-09-02 08:05
导读:本文分享了OGD对BCS I类药物的BE豁免监管经验,特别是抗肿瘤药物。
1、引言
仿制药活性成分的吸收速率和程度与参比制剂相同则两种药品是生物等效的(21 CFR 320.1(e) and 320.23(b))。人体生物等效性(BE)研究是确保仿制药与参比制剂(RLD)等效的优选方法。体内BE研究需要在制药、伦理、临床、药代动力学、分析和统计方面精心设计。实际上通常难以招募患者进行体内研究,如病例少或病情发展快的癌症。可用体外试验代替BE研究来证明仿制药与参比制剂生物等效。
FDA应用BCS的原理将体外试验作为口服速释制剂BE豁免的一个工具。此法的原理是药物的溶解性和渗透性及药品的溶出度这三个因素决定了口服药物在吸收部位的吸收速度和程度。BCS I类药物具有高溶解性和高渗透性,如果将BCS I类的药物制成相对于胃排空快的速释制剂,且不含影响药物吸收和胃肠道转运的赋形剂,则可豁免BE研究。
FDA于1995年开始实施BCS模式作为监管工具,当时发布了速释制剂(IR)放大生产和批准后变更(SUPAC)指南,在该指南中介绍了高溶解性和高渗透性药物的监管。1995年至2000年生物豁免仅限于该药品的放大或其他变更的补充。2000年8月FDA发布BCS指南,扩大了对含有BCS I类药物的口服速释制剂的监管考虑。FDA在该指南中进一步阐述了高渗透性药物的定义。
含BCS I类的药品,仿制药办公室(OGD)鼓励申请人申请豁免体内BE研究,并提供关于药物高溶解性和高渗透性、药物在胃肠道中的稳定性和药物快速溶出的相关文件,详见BCS指南。OGD已经审查和批准了几种抗多源性肿瘤药物基于BCS的BE豁免申请。对于抗肿瘤药物,考虑到进行体内BE研究的复杂性,基于BCS的BE豁免具有许多优点。对于具有细胞毒性的抗肿瘤药物也是如此。通常在健康志愿者体内进行细胞毒性药物的研究是不道德的。21 CFR 320.31文件指出仿制药研究企业在进行BE研究之前需要提交一份临床新药(IND)申请审查(也称为Bio-IND)。如果人体内研究设计不安全,OGD在审查Bio-IND后出于安全考虑会让临床试验暂停。抗肿瘤药物的研究需要在多个临床试验点进行,以便招募足够数量的患者。此研究受试者之间的变异性较高,不良事件较多,甚至会有患者死亡。
本文分享了OGD对BCS I类药物的BE豁免监管经验,特别是抗肿瘤药物。讨论了这类药物推迟批准的问题。基于BCS的BE豁免除了降低研发成本和缩短研发时间外,预计还可以缩短审查和批准的时间来减少监管负担,但是很多申请人似乎错过了机会。因此把出现的问题拿出来便于降低抗癌药物的仿制成本和保证治疗效果等效以便于尽快获得批准。
2、方法
本文对过去10年提交的新药申请(NDAs)和简略新药申请(ANDAs)进行了调查,其中包括对BCS I类的豁免要求。包括多种适应症的药物,如治疗癫痫、癌症、焦虑、痴呆、恶心、呕吐等药物。根据对BCS I类数据的审查,确定了药物评估和研究中心(CDER)认定的至少有4种不同的药物有资格获批基于BCS的BE豁免。还发现一种用于治疗癌症的药物,其仿制药申请者已经申请了基于BCS的BE豁免,但由于信息不足,申请的BE豁免尚未定论。这五种仿制药,一个ANDA是基于BCS豁免的,而另一些则进行了人体内BE研究(即基于非BCS的)。我们评估了每个申请的审评周期,确定了这些研究中的常见缺陷,并比较了基于BCS和基于非BCS的ANDAs之间的审评过程。
3、结果与讨论
CDER审评了提交的数据评估了55种口服速释固体制剂的数据,55种药物中的41种产品符合基于BCS的BE豁免要求。41种药物中有4种(约10%)是抗癌药,相比之下,WHO的基本药物清单中有21种为BCS I类,只有1种是抗癌药物。
迄今为止,OGD已经通过了我们的调查确定的4种抗癌药物的ANDAs。在这49份申请中,30份ANDAs(60%)有体内的BE研究数据,数据显示在禁食或进食条件下和参比制剂等效。其他19个ANDAs(40%)的申请人偏向基于BCS的BE豁免方法,提交了3种体外检测数据:渗透性、溶解性和溶出。
如果ANDA申请人在提交的文件中提供了足够的信息和数据支持仿制药的生物等效性,那在一个审评周期内,FDA可以在生物等效性要求方面接受申请。但是如果提交的信息不充分或者数据不足以支持生物等效性,FDA会向申请人寄出数据不足的回复信,并要求提供更多的数据信息,这会导致多周期审评和延迟批准申请。
对于有体内BE研究的30个药品申请,23个ANDAs已评估完,并且这23个ANDA中的11个的生物等效性研究可以接受,仅需要一个BE审评周期。这占了这一类申请总数的将近一半(48%)。相比之下,包含体外BCS相关研究和基于BCS的BE豁免的19个申请,其中11个ANDAs已审评完,只有1个ANDA(占已审评总申请的9%)包含完整、合适的BCS相关信息且能在一个审评周期内完成BE部分的审评。而其他10个ANDAs经历了2~5个审评周期。
下面讨论的案例中可以找到与具有体内BE研究的药物审评周期(48%在第一个审评周期内即可接受)相比大部分基于BCS的BE豁免申请审评周期(在第一个审评周期内可接受的不足10%)较长的部分原因。每个案例都涉及不同的药物。下面的案例研究突出了审评不同的ANDAs时发现的主要问题。
案例1:
基于BCS I类抗肿瘤药物的BE豁免。该药为前体药物,在体内转化为活性代谢产物后发挥作用,其渗透性可能会受转化机制和转化部位影响。由于缺乏该药物在体内的代谢转化的位置及机制的具体信息,无法根据体外试验数据批准前体药物的BE豁免申请。FDA的BCS指南描述了对前体药物和活性代谢物进行体外研究的注意事项,因此该申请人应再提交所有活性代谢物的体外试验数据。
案例2:
基于BCS的细胞毒性的仿制药的BE豁免申请,申请人仅提交了体外溶出数据支持BE豁免申请。申请人仅靠已发表的文献支持药物具有高溶解性和高渗透性,而未进行溶解性和渗透性研究。审评中心不接受采用已发表的文献作为基于BCS的BE豁免证据的申请。文献中可能不含测试机构对研究做出判断的必要细节,而无法验证同行的文献,因此审评中心建议申请人提供溶解性和渗透性的研究数据,详见CDER工业指南。
案例3:
申请人引用了RLD标签中的信息作为高渗透性的证据,且提交了体外试验数据支持该药为高溶解性和药品快速溶出。稳定性试验数据显示该药在胃肠道的某些pH条件下不稳定。为了达到基于BCS的BE豁免的目的,FDA认为药物体外数据证明药物在胃肠道中稳定且药物在人体的吸收率至少达到90%,则可认为该药具有高渗透性。审评中心认为虽然药物在胃肠道某一pH值时不稳定,但申请人未在提交的文件中提供证据证明符合基于BCS的BE豁免的要求。
为了获得基于BCS的BE豁免资格,审评中心需要审评药品在胃肠道中不同pH环境中是否能快速溶出。此时,申请人提供在多种溶出介质(0.1 N HCl溶液、pH4.5缓冲溶液和pH6.8缓冲溶液)中进行溶出测试得出数据,该药能在一些pH溶液中快速溶出,但不是在所有pH值的介质中快速溶出。由于药物制剂中的辅料在低转速时引起锥体效应,导致药物不能完全溶出,审评中心要求申请人使用指南中建议的USP装置开发一种更合适的溶出方法证明该药品能快速溶出。
除了上述案例中讨论的主要缺陷之外,还在这些案例中发现了其他常见的缺陷。这些缺陷主要与提交的资料不完整有关,如果多加注意就可以避免这些缺陷。按照研究类型分类,将这些缺陷总结在表1中。
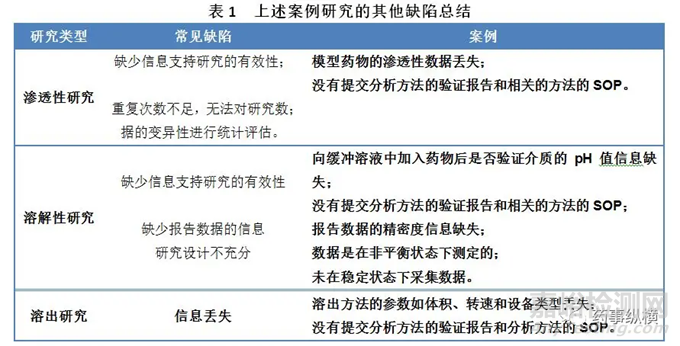
以上三个案例反映了基于BCS的BE豁免申请与ANDA申请一起提交时反复出现的问题。
虽然这些案例研究中的问题是在对抗肿瘤药物的审评时发现的,但是OGD的经验表明这些问题不只出现在特定的某一类药物中,一般适用于将体外渗透性、溶解性和溶出曲线测试应用到基于BCS I类的BE豁免仿制药。大多数批准时间推迟与数据存在问题(高变异性、重复性不好)、检测方法不合适、缺乏研究的支持信息(如溶解度、渗透性和溶出测定方法以及药物或活性代谢物定量分析方法的方法学验证报告)有关。
上述案例在申请基于BCS的BE豁免建立生物等效性时突出了一些需要考虑的因素,如:前药的转化位点是在肠道还是在肠道吸收以后的其他部位,可能需要对活性代谢物进行体外渗透性研究。前药和药物的溶解性数据也是非常重要的。这些信息应与申请文件一起提交并且应避免修改申请。当数据不支持药物的稳定性或药物在所有pH条件下快速溶出亦或药物以被动扩散的方式通过渗透膜时,申请人应该考虑为什么ANDA仍然应该考虑基于BCS的BE豁免。
相比之下,申请人进行抗肿瘤药物体内BE试验的挑战主要是临床试验设计和实施,因为细胞毒性和其他具有严重毒性的药物的BE研究一般在患者体内进行而不是在健康志愿者体内进行。研究方案需要确保试验设计充分和患者的安全。FDA法规要求申请人在开始进行细胞毒性药物的BE研究之前需要提交Bio-IND,并对提交的Bio-IND试验方案进行审评和提出指导意见。FDA还对提交的非细胞毒性药物的研究方案提出了意见和指导。对于案例2和3中确定的原料药,FDA已经审评了几个研究方案(其中一些是在Bio-INDs中提交的),并对研究方案的关键点进行了指导,包括以下内容:
(1)试验对象选择
由于严重的细胞毒性,建议在患者体内进行研究。试验人群应与创新药的标签中描述目标患者人群及适应症相对应。由于很难招募目标患者,因此申请人可以在多个临床机构和多组进行研究。
(2)剂量选择
为了让患者参与研究而修改或中断患者的治疗剂量方案是不道德的,因此研究建议的剂量对于患者的个体给药方案是特定的。不建议对此进行标准化要求,但患者可以在一个剂量范围内选择。每个患者在所有研究时间内接受的相同的剂量,以避免难以解释研究数据。
(3)给药方案、采样时间和清除期
在药物标签中描述的治疗指南应对给药方案提出建议,还应考虑药物的消除速率和半衰期等药代动力学参数以确保选择合适的采样时间和清除期。
(4)空腹或进食
这些建议是基于RLD标签中的描述。一般来说OGD建议在空腹和进食条件下进行BE研究,但是有时候由于安全问题而妨碍了在这两种条件下进行BE研究。对于肿瘤患者预计会伴有高血压、糖尿病或高胆固醇等疾病,让这些患者食用高脂肪食物后进行治疗研究是危险的或不道德的。并且厌食症患者可能不会将全部食物吃完而导致整体数据难以解释,因此OGD同意让患者食用改性膳食物后进行研究。
(5)合用药物
建议不要与非研究药物一起服用,应考虑非研究性药物的药理学及药代动力学可能会降低研究药物的清除率,并且药物之间可能相互作用。
(6)统计学考虑
如果在多个试验点或多个分组进行研究,则建议在统计模型中测试这些变量带来的影响。
与上述基于BCS的BE豁免的ANDAs相比,非基于BCS的ANDAs缺陷相对较少,可能是在审评Bio-INDs之后(进行临床研究之前)审评中心提出了建议。
我们调研了与上述案例具有相同药物的申请,确定了体内研究的缺陷。与案例1相关的药物ANDAs中没有进行BE研究。OGD已经审评了至少5个ANDAs,其中包括与案例2和案例3有关的药物体内BE研究数据。其中两个ANDAs的BE研究在第一审查周期内就可以通过,其他三个ANDAs存在缺陷但也是可忽略的,这三个ANDAs中的两个药物的缺陷:一个是分析报告中缺少信息和一个与临床报告有关。
经验表明,在一个审查期内,大多数非基于BCS的ANDAs的BE研究足够充分,这些ANDAs的审批时间较短(提交ANDAs之前和Bio-INDs的时间也考率在内)。
4、结论
基于BCS的BE豁免可以免做生物等效性研究,要求药品是符合指南中的某些标准的速释制剂。只有在审评申请材料的数据之后FDA才能对BE豁免的申请作出决定。FDA的BCS指导原则旨在帮助速释口服固体制剂的申请人豁免BA或BE研究。希望以上案例可以帮助申请人避免上述案例中的问题,根据指南的要求进行研究并提交完整的高质量的申请资料。

来源:Internet