
嘉峪检测网 2024-09-29 08:31
导读:本文以化学药品为例,从审评工作者的角度,分析当前我国药品生产场地变更的监管背景和工作中的常见问题,提出对应管理建议和研究思路。
[摘要]药品作为特殊商品,其质量与原辅料来源、产品生产工艺和控制条件、企业质量管理水平息息相关。药品生产场地变更作为药品上市后变更的常见情形,常伴随着上述要素的改变,给药品质量带来不同程度的影响和风险,加强药品生产场地变更管理和研究,确保变更后药品安全、有效和质量可控,是药品上市许可持有人和监管部门的重要责任。本文以化学药品为例,从审评工作者的角度,分析当前我国药品生产场地变更的监管背景和工作中的常见问题,提出对应管理建议和研究思路。
药品生产场地与药品质量密切相关[1],“药品集中采购”“一致性评价”等医药政策的持续完善和行业发展因素,境内药品生产企业药品生产场地变更需求不断上升,《药品管理法》《药品注册管理办法》《药品生产监督管理办法》及相应配套文件的深入实施,我国药品监管科学水平的不断提升对监管机构提出了更高要求和挑战。亟需细化药品生产场地变更管理要求和技术要求,降低政府监管风险的同时赋予药品上市许可持有人(以下简称“持有人”)更大的经营灵活性,促进产业优势聚集和高质量发展。
1、 监管政策背景
2019年新的《药品管理法》及持有人制度实施以前,对药品生产场地的变更管理没有统一的法规要求,涉及有三种情形:一是批文转让(药品技术转让)关联发生的生产场地变更,执行《药品技术转让注册管理规定》(国食药监注[2009]518号);二是委托生产关联发生的生产场地变更,执行《委托生产监督管理规定》(2014年第36号);三是药品生产企业内部改变药品生产场地关联发生的生产场地变更,执行原《药品注册管理办法》及食药监药化管[2015]122号文件。
以上三种情形的药品生产场地变更分属于不同法律文件管理,审评审批主体不同[2],工作程序和技术要求散见于各文件中,在具体操作层面,同一风险级别的生产场地变更,研究要求都可能存在差异,部分规定甚至限制了药品生产技术的合理转移,不利于政府监管和企业申报变更[3]。原国家食药监总局曾拟定了药品生产场地审批管理专门文件,2017年、2018年两轮征求意见后未正式出台[3-4]。
新的《药品管理法》实施后,药品生产过程中的变更由原来的按变更内容分类管理,修订为按变更的风险程度分类管理(重大、中等和微小)。《药品管理法》及《药品注册管理办法》要求持有人全面评估、验证变更事项对药品安全性、有效性和质量可控性的影响,更加强调风险管理和全程管控[5]。
2021年实施的《药品上市后变更管理办法(试行)》从内容上将药品上市后变更分为:持有人变更、生产场地变更和其他事项变更。生产场地变更进行了单列和专述,持有人变更往往也关联发生药品生产地址的变更(即原来的药品技术转让)。国家未单独制定药品生产场地变更的管理规定,但生产场地变更是药品上市后变更的最常见情形之一,在药品全生命周期监管工作中占据重要比重。
《药品上市后变更管理办法(试行)》对场地变更的“定义”“审批事权”及相应管理要求进行了明确(见表1),但缺乏基于风险的差异化管理措施和研究要求。

2 “药品生产场地”及其变更的实质和内涵
药品生产管理的传统通常将“药品生产场地变更”理解为“药品注册批件所载生产地址的改变”[3],忽略了注册批件所载地址不变但实际物理地址和空间改变的情形。生产场地变更过程中,药品生产环境、操作人员、设施设备、文件系统、质量管理体系等都可能发生关联变更并给药品质量带来负面影响,管理工作理应考虑。
2020年7月31日发布的《药品上市后变更管理办法(试行)(征求意见稿)》中对生产场地变更概念的阐述较正式稿更为详细:“药品生产场地变更是指药品生产许可证、药品批准证明文件载明的生产场地(含自有生产场地、受托方生产场地)地址、位置和生产单位的变更,其包括原址或者异地迁建、新建、改建和扩建车间或生产线”。再参考2017年发布的《药品生产场地变更研究技术指导原则(征求意见稿)》对生产场地的定义以及龚青等人对药品生产场所和生产线的阐述[6],笔者认为药品生产场地可定义为“为满足药品生产要求的一系列生产要素组合,涵盖了药品生产环境、硬件、软件等质量管理体系要素”,实质代表了持有人保障药品生产活动持续、稳定、可控的能力,内容远远大于“生产车间”“生产线”的概念。对其变更的管理应以不影响药品安全、有效和质量可控为评价指标,以不干扰药品生产活动的持续、稳定、可控为原则。
不同类别药品的“药品生产场地变更”风险也不尽相同,化学药品生产场地变更有其自身特点,应区分风险高低,合理分配监管资源[7]。应将“质量管理体系和要素是否发生系统性、整体性变更;对产品生产行为、生产工艺是否产生系统影响;是否有较大药品质量风险”作为评价标准,经评估后采取与之对应的风险管理路径和技术研究策略。
3、 药品生产场地变更管理研究
结合我国法规对不同类型药品生产场地变更进行分析和风险评估(见表2)。
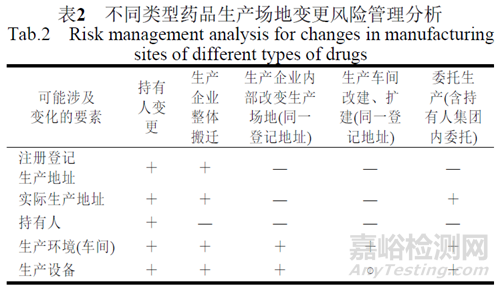
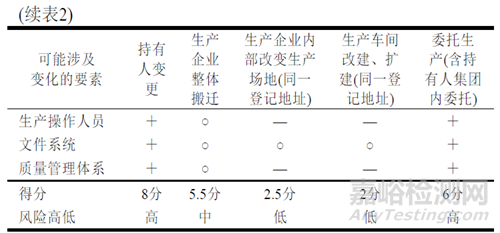
注:通常发生变更的为“+”,计1分,部分发生变更或不确定的为“○”,计0.5分,不发生变更的为“-”,计0分;6~8分为高风险,3~6分为中分析,0~3分为低风险。以常规制剂分析,未考虑具体品种、剂型、生产工艺和变更程度等,若生产无菌制剂、有毒或高活性产品以及生产品种、剂型较多的企业,应增加分值;外用制剂或生产品种较少的企业可降低分值;持有人变更可能发生整厂转让,生产地址和操作人员不变的情形,分值对应降低;变更包装工序等场所,其分值也相对较低。具体可参照ICH Q8附录1和附录2开展评估[8]。
基于以上分析,不同类型的生产场地变更风险差异大,如新法规实施前按备案管理的委托生产事项,在几种变更情形中风险相对较大,持有人变更关联发生的场地变更,应区分质量要素变更情况后处理。
4、 化学药品制剂生产场地变更技术审评常见问题
4.1 变更过程技术考量
(1)充分、系统的质量风险评估,持有人应从“人、机、料、法、环”等因素着手,对影响化学药品制剂质量的生产环境、设备、工艺参数、原辅料来源、批量、检验数据可靠性等进行风险识别、评估和控制[8],制定项目实施方案,项目管理手段的应用有助于变更的顺利实施并通过监管部门审批。(2)减少可变因素,《已上市化学药品药学变更研究技术指导原则(试行)》(以下简称《指导原则》)要求新旧场地处方、生产工艺、批量等应保持一致,实际生产过程中不一定能完全做到,但减少可变因素有利于提高场地变更成功率。(3)确保变更前后“质量等同”,一般来说,已上市药品(即变更前产品)的质量是经过验证和监管部门认可的,药品生产场地变更应围绕变更后产品与变更前产品质量等同原则开展[9]。(4)基于质量源于设计的理念以及ICH 关于药品质量的定义和要求,药品关键质量属性(CQAs)依赖于生产过程中的关键物料属性(CMAs)和关键工艺参数(CPPs)的控制,并参照质量标准对产品进行检验及稳定性研究,应从处方工艺控制、质量对比研究、稳定性对比研究三方面确保化学药品制剂变更前后“质量等同”(见表3)[10]

4.2 技术审评常见问题分析
(1)科学严格的处方工艺过程控制,是保障药品质量的前提。持有人处方工艺研究常见问题有:①某液体制剂配制过程中,药液需从A 罐转移到B 罐,未考虑变更前后管路差异带来的处方投料量损耗;②含热敏成分的产品,干燥设备显示温度与物料温度存在差异,仅对比了显示温度,或箱式干燥变为沸腾干燥,仅保证干燥操作时间一致,实际变更后物料由于进出先后不同存在受热差异;③散剂产品变更后批量增大,包装时间延长,未考察水分变化情况,无菌制剂变更后灌装时间延长,无菌风险增大;④小剂量药物未参照《化药口服固体制剂混合均匀度和中控剂量单位均匀度研究技术指导原则(试行)》对混合工序进行研究;⑤原辅包发生关联变更,未进行相应的对比研究(如原料药生产工艺对比、难溶性原料药的晶型或粒度研究、辅料含水量检测、包材匹配性及密封性研究等);⑥生产工艺涉及中等以上变更,持有人未按《药品注册管理办法》提交备案或补充申请。
(2)质量研究可以客观、全面及灵敏地反映产品质量变化情况,是验证生产场地变更前后药品质量一致、等同的重要手段[11]。常见问题有:①产品本身质量控制水平不高(如杂质控制项目偏少或缺失),仅依据质量标准开展对比研究,研究项目不充分,或所选的检测项目或指标不具代表性(如未按照《指导原则》开展溶出曲线对比研究);②变更前后检验数据存在明显差异或异常,未进行充分的调查评估(如OOS、OOT 调查),分析方法转移研究不完善;③杂质检测结果有变化,但未结合《化学药物杂质研究的技术指导原则》及每日允许暴露量(PDE)开展安全性分析评估;④参比品选择不合理,未选择国家公布的参比品或市面上安全性数据充分的产品;⑤批量的选择未遵循《化学仿制药注册批生产规模的一般性要求(试行)》,或研究批次产品检验结果与日常产品生产情况不吻合。
(3)生产场地变更后,可能造成产品稳定性的变化,《指导原则》要求对变更后1~3 批样品进行加速和长期稳定性研究,申报时提供3~6 个月稳定性研究数据,持有人可参照《化学药物(原料药和制剂)稳定性研究技术指导原则(修订)》和《中国药典》2020 版9001《原料药物与制剂稳定性试验指导原则》开展产品稳定性研究。常见问题有:①考察项目不全面,如未对有关物质、杂质等进行考察,大容量注射液等采用半渗透性容器包装的产品未开展失水率考察或考察方法不合理,西林瓶包装产品未考察倒置或水平放置情况;②数据分析不完整,如有关物质、不溶性微粒等项目仅汇总检测结论,未比较各考察时间点的实测值,或变更前后部分数据出现异常结果及趋势,未开展分析评价;③未严格执行稳定性试验方案,如由于企业检验或生产经营影响,取样时间和取样频率有差异(半月以上);④提交的数据、图谱与实际有误差。
5、 结语
监管部门应结合《药品生产监督管理办法》关于“场地管理文件”的要求进一步明确“药品生产场地”基本定义和内涵。鉴于药品生产场地变更的复杂性以及与常规药品上市后变更的差异性,应探索基于风险的、针对不同变更类型的分类审批及监管措施以强化风险管控,节约监管资源。应配套技术文件指导持有人研究,其中2017 年发布的《药品生产场地变更研究技术指导原则(征求意见稿)》仍值得借鉴。
持有人应重视项目管理技术在项目立项、方案制定和推进过程中的运用,注重前期策划、中期控制、后期总结完善,如变更前长期未生产,相关数据不完善或处方工艺不合理的,应评估项目可行性;关注时间逻辑可避免重复试验或缺陷数据产生,如完成原辅包研究后再开展批量化生产研究,完成设备确认后再开展工艺验证;稳定性试验期间,可提前进行初期数据对比和分析以节约研发申报周期。“保持变更前后产品质量等同”是变更的核心原则,变更过程中应尽量减少处方工艺因素的干扰,关注研究用样品选择合理性,重点证明变更前后产品杂质谱、关键理化性质一致,其中杂质研究是化学药品场地变更研究的重要内容,可参照国内外药品监管机构、药典等建立合适的分析方法,进行系统的杂质谱分析,重点关注变更后产生的降解杂质及产品稳定性改变。《指导原则》要求的1~3批检验批量和研究批量,以及3~6 个月稳定性研究数据,仅是对于提交资料的最低要求,实际应针对产品情况开展更为全面的研究,对变更后的质量数据及稳定性结果进行跟踪分析,以便顺利推进变更并保障药品质量。
参考文献
[1]赵兰婷,马玲玲,卢雪明,等.已上市中药制剂生产场地变更管理现状分析[J].中国医药导刊,2023,25(12):1219-1222.
[2]谢金平,孙圆圆,彭楠,等.药品上市许可持有人(MAH)制度对现行监管制度的影响及衔接建议[J].中国卫生政策研究,2018,11(12):1-6.
[3]国家食品药品监督管理总局办公厅.总局办公厅公开征求《药品生产场地变更简化注册审批管理规定(征求意见稿)》及《药品生产场地变更研究技术指导原则(征求意见稿)》意见[EB/OL].(2017-10-12)[2024-05-08]。https://www.nmpa.gov.cn/xxgk/zhqyj/zhqyjyp/20171017170901778.html.
[4]国家食品药品监督管理总局.总局关于公开征求《药品生产场地变更注册审批管理规定( 征求意见稿) 》意见的通知[EB/OL] . (2018-3-21)[2024-05-08].https://www.nmpa.gov.cn/xxgk/zhqyj/zhqyjyp/20171017170901778.html.
[5]夏东胜,田春华,王涛.从《药品管理法》制修订解析我国药品监管理念变化趋势[J].中国药物警戒,2021,18(09):837-844.
[6]龚青,田洁.我国化学药品申报资料中关于生产线相关要求的审评考虑[J].中国药学杂志,2020,55(03):239-244.
[7]李晓宇,杨悦.美国化学药品生产场地变更的法规研究[J].中国药学杂志,2016,51(08):671-677.
[8]ICH . Pharmaceutical Development (Q8(R2)) [EB/OL] . (2009-08-01)[2024-05-08].https://database.ich.org/sites/default/files/Q8%28R2%29%20Guideline.pdf.
[9]国家药品监督管理局药品审评中心.《已上市化学药品药学变更研究技术指导原则( 试行) 》[EB/OL] . (2021-02-10)[2024-05-08] . https://www.cde.org.cn/main/news/viewInfoCommon/4ec3dca752a82347bdf24ad3d3e85113.
[10]许静玉,唐谦,钱璟,等.化学药品口服固体制剂生产场地变更的技术风险考量[J].中国现代应用药学,2022,39(05):690-694.
[11]王筱婧,王东兴,祝倩,等.药品技术转让质量对比研究的主要内容与关注要点[J].中国医药科学,2016,6(14):221-223.
内容来源:广东化工 2024年第11期

来源:Internet
关键词: 药品