
嘉峪检测网 2024-11-06 15:23
导读:本研究通过问卷调研与现场实地调研相结合的方式对附录的实施情况进行调查,发现企业在实施过程中存在的问题与困难,为附录的有效实施提供参考建议。
摘要 目的:调研我国GMP临床试验用药品附录(简称附录)自发布以来的实施现状,了解企业执行过程中存在的问题,为附录的有效实施提供建议。方法:采用线上问卷与现场调研相结合的方式对不同省份企业的实施情况进行归纳,分析企业对相关要求的执行情况及存在的问题。结果:参与调研的企业共100家,来自10个省份,产品覆盖面广且具有代表性。调研显示,大部分企业能够符合附录的要求,但仍有部分企业在质量管理体系的建立(8%)、共线评估(9%)、临床试验用药品档案的建立(15%)、对照药品与试验用药品的相似性评价(10%)、留样(24%)、放行(21%)方面存在不足。结论:建议申请人切实履行主体责任,贯彻附录要求,组织更多专业性培训提升人员综合水平;药监部门发布相关的指导性文件,基于风险对临床试验用药品进行专项检查或抽样检验,促进附录要求落实落地。
临床试验用药品与已上市药品相比,往往具有生产工艺不成熟,特性、潜在作用及毒性了解不充分,关键质量属性识别不充分,检测方法研究不完善等特点。其制备过程较为复杂,涉及试验用药品与对照药品(包括安慰剂或已上市药品)的同时制备,有时可能涉及盲法操作,对包装、贴签等带来更多挑战。
为规范临床试验用药品的生产,不同国家或组织相继发布了临床试验用药品生产质量管理规范[1]。美国FDA于1991年发布了《新药临床试验用样品技术指导原则》[2],于2008年发布了《Ⅰ期临床试验用样品生产质量管理规范》[3],Ⅱ期和Ⅲ期临床试验用药品遵循1991年发布的指导原则与美国联邦法规21 CFR(Code of Federal Regulations)第210部分与211部分的要求;欧盟现行版药品生产质量管理规范(Good Manufacturing Practice,GMP)《临床试验用药品的生产》附录为2017年发布[4];我国于2022年5月正式发布GMP《临床试验用药品(试行)》附录(以下简称“附录”),并自2022年7月1日起施行[5]。
临床试验用药品的生产处于药品研制环节,企业通常存在不能根据研发进度及时跟进各环节法规要求的情况[6],此外临床试验用药品生产的检查是在药品注册环节开展的回顾性检查,检查的及时性、系统性有所欠缺。附录发布以来,附录在企业中的执行情况尚缺乏有效的了解。本研究通过问卷调研与现场实地调研相结合的方式对附录的实施情况进行调查,发现企业在实施过程中存在的问题与困难,为附录的有效实施提供参考建议。
1、 临床试验用药品附录概况
附录共包括十四个章节,五十一项条款,内容重点强调了临床试验用药品与商业化药品存在差异的地方(见表1)。

2、调研基本情况
根据附录的基本要求,结合专家意见,调研小组讨论设计了调研问卷。问卷内容主要包括临床试验用药品企业质量管理体系的建立情况;制备车间的GMP符合性情况及共线生产情况;临床试验用药品档案的建立情况及其各项内容的实施情况;制备过程中的相关验证或确认情况、盲法试验中包装与贴签的管理情况;临床试验用药品的留样管理情况;产品放行时质量评价的内容实施情况。
调研共收到有效问卷98份,问卷填写人员职位主要包括注册经理、研发负责人、生产负责人、质量负责人、副总裁、法规部总监、临床运营专员等。根据书面调研情况对2家企业进行了现场实地调研。调研企业共计100家,涉及我国山东省(39%)、江苏省(15%)、重庆市(9%)、辽宁省(8%)、广东省(5%)、河北省(10%)、福建省(3%)、上海市(6%)、四川省(2%)、浙江省(3%)共10个省份,企业分布范围广。
被调研企业包括民营企业、国有企业、国有控股企业、中外合资企业、外资企业、股份制企业等各种类型;既包括300人以上的大型企业(占比63%),也包括中小型企业;产品类型包括化药(企业占比68%)、中药(企业占比7%)、生物制品(企业占比35%),部分企业存在生产多种类型产品的情况,药品种类覆盖面广,具有代表性。
3、 调研结果及分析
3.1 质量管理
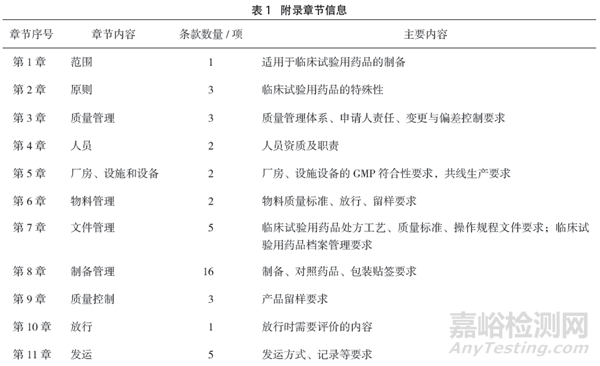
附录要求,临床试验用药品制备单位应当基于风险建立质量管理体系。此部分调研了企业建立有效运行的质量管理体系(进行了质量风险管理,建立了变更、偏差、自检等质量管理制度,并按规定进行了管理和记录)的时间阶段。数据显示,不同企业根据不同产品需要开展的临床试验情况可能会在不同的阶段建立有效的质量管理体系,但参与调研的100家企业中还有8%在临床试验用药品制备阶段并未建立有效运行的质量管理体系,而是在临床试验完成后的上市前阶段才开始建立(见图1)。
3.2 厂房、设施和设备
附录要求制备临床试验用药品的厂房、设施和设备应当符合GMP及相关附录的基本要求,共线生产情况下,应当进行共线可行性评估,并采取适当措施控制污染与交叉污染的风险。
本次对临床试验用药品制备车间的GMP符合性情况进行了调研,数据显示,几乎所有的临床试验用药品都是在不同GMP符合程度的生产车间中制备的,其中约80%的制备车间已经通过了GMP符合性检查,其余制备车间虽然还未进行GMP符合性检查,但是经企业内审或第三方审计符合GMP要求。
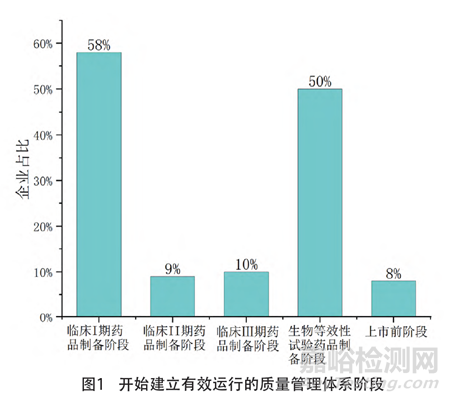
87家企业存在共线生产情况,79家(91%)进行了共线生产风险评估且评估包括了所有共线的产品,8家(9%)未进行共线评估或评估中仅包括商业化产品而未包括临床试验用药品。共线评估企业中大部分根据《药品共线生产质量风险管理指南》的要求,考虑了拟共线生产品种的特性、工艺、预定用途、厂房设施设备共用情况等因素[7](见图2)。
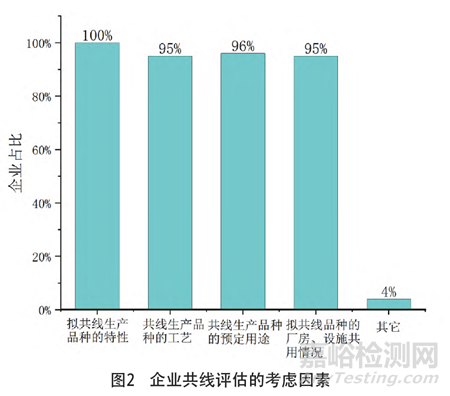
在共用厂房、设施或设备情况下,大部分企业采用阶段性生产、不同品种转产时进行清洁确认或验证的方法控制污染或交叉污染风险,有的企业采用密闭系统、划区域隔离、与产品直接接触的部件为专用或一次性系统等方法(见图3)。
3.3 文件管理
附录要求申请人应当建立临床试验用药品档案,并随药物的研发进展持续更新,且对档案的内容进行了规定。
参与调研的企业中共9 6 家申请人, 8 2 家(85%)已建立了临床试验用药品档案,14家(15%)的申请人还未建立。已建立档案的企业中,各项内容实施情况见表2。

数据显示,除历次成品标签占比略低(72%)外,其余内容占比均在85%以上,这说明,在已建立临床药品档案的企业中,绝大多数档案具备了附录所要求的基本内容,但是部分企业(28%)容易忽略历次成品标签项目。
3.4 制备管理
3.4.1 制备
附录要求应当制定清洁操作规程明确清洁方法,并进行必要地确认或验证;无菌药品的灭菌工艺或无菌生产工艺应当遵循现行相关技术要求,确保其无菌保证水平;生物制品应当确保病毒等病原体或其他外源因子灭活/去除效果。
随着临床试验阶段的推进,企业的验证工作会逐渐趋向完善。本次重点调研了早期临床(Ⅰ期)样品制备阶段或生物等效性试验样品制备阶段清洁验证、无菌验证及生物制品病毒灭活/去除验证的执行情况(见表3)。

数据显示,91%的企业对关键或所有设施设备进行了清洁确认或验证;涉及无菌生产工艺的企业中,共96%的企业进行了不同程度的无菌模拟验证;所有适用企业均进行了病毒去除灭活验证。数据表明,在早期临床或生物等效性试验样品制备阶段,大部分企业已经开展了不同程度的清洁、无菌、病毒去除相关的确认或验证工作。
3.4.2 包装贴签
附录要求,如果涉及盲法试验,应当对包装的外观相似性和其他特征相似性进行检查并记录;需要将对照药品改变包装或标签时,应当评估并证明所进行的操作未对原产品质量产生明显影响;应当采取有效措施防止试验药物与对照药品出现贴签错误。
参与调研的企业中涉及盲法试验的共60家,90%(54家)进行了相似性评价,评价指标一般包括包装、标签、剂型、外观、颜色、味道、气味等,10%未进行相似性评价;共41家企业存在对照药品改变包装或标签的情况,其中95%(39家)评估了所进行的操作对原产品的影响,评估中的关注点包括稳定性、溶出度、纯度、杂质、效价/活性、外观、含量、有效期等,5%未评估改变包装或标签的操作对原产品的影响;生产过程中为防止试验药物与对照药物出现贴签错误采取的措施包括进行标签数量平衡计算与清场、由经过培训的人员进行中间控制检查、对照药物与试验药品不在同一包装线包装或错时包装等。
3.5 质量控制
附录要求,每批临床试验用药品均应当留样,留样应当包括试验药物和安慰剂,数量为2倍全检量;已上市对照药品可基于风险原则确定留样数量;留样应当包括已设盲的试验药物、对照药品(含安慰剂)。
参与调研的企业中去除1家不负责留样的受托企业,其余99家企业中85%对试验药品进行了留样且留样数量满足2倍全检量;使用安慰剂的企业共63家,其中87%对安慰剂进行了留样且满足2倍全检量要求;涉及盲法试验的企业共61家,其中85%留样包括了已设盲的试验药物与对照药品(含安慰剂),见表4。综合上述各种留样不规范的情形,且去除重复统计的企业,共24家企业(24%)未按要求进行留样。

3.6 放行
产品放行时需进行质量评价的内容、每项内容的实施企业数量及执行了此项内容的企业占比见表5。参与调研的企业中,去除4家受托企业,96家申请人产品放行时,企业需要评价的基本内容中“贮存条件”占比略低,为79%,其余占比均在90%以上。这说明,部分企业(21%)在放行时容易忽略贮存条件因素。

4 讨论与建议
4.1 建议申请人切实履行主体责任,贯彻执行附录要求
附录要求申请人对临床试验用药品的质量承担责任。对于自主生产的产品,申请人应当对自己在质量管理方面的现状与我国附录要求之间存在的差距加以识别,不断提升质量管理水平;对于委托生产的情况,少数被调研企业未对受托企业进行质量审计,大部分企业虽然进行了质量审计,但是很多企业反映,审计过程中存在受托企业的部分现场不接受审计、申请人与受托方质量管理理念存在差异、申请人与受托方质量体系难以衔接、受托单位不配合提供完整资料等问题,建议申请人与受托方之间加强沟通与合作,通过签订质量协议进一步明确双方责任,保障临床试验用药品的安全、有效、可控[8-11]。
4.2 建议组织专业性培训,提升企业人员对附录的理解水平
参与调研的部分企业反映,还未对附录进行过系统的学习,调研数据中8%的企业在临床试验用药品制备阶段未建立有效运行的质量管理体系,15%的企业还未建立药品档案,这些问题也能够说明部分企业缺乏临床试验用药品的生产质量管理意识,对附录的内容理解还不到位。这需要企业与药监部门进一步加强对附录的培训,以加深企业人员对附录的理解和认识;需要针对临床试验用药品的特殊性,加强对相关人员的专业知识培训,提高制备人员的综合素质。
4.3 建议增加指导性文件,指导企业具体实施
临床试验用药品的研制是一个从不成熟到逐渐成熟的过程,参与调研的部分企业反映对不同临床试验阶段的确认与验证程度、生产控制程度、质量控制程度等难以把握。美国FDA有专门针对Ⅰ期药品制备的指南,并明确了Ⅱ期、Ⅲ期药品制备应当遵循的规定[3];WHO临床试验用药品GMP指南中也有对不同临床试验阶段的要求,如“早期临床中可以使用与最终剂型差异很大的剂型,但是在Ⅲ期临床研究中,剂型应与商业化规格相似”“Ⅲ期临床的剂型应按商业化生产药品要求进行表征,得到相同水平的保障”[12]。建议我国根据临床试验不同阶段药品制备的特点与重点,增加相关的指导性文件,以指导企业具体实施。
4.4 建议监管部门基于风险开展临床试验用药品专项检查或抽样检验
美国《Ⅰ期临床试验用样品生产质量管理规范》规定:“在某些状况下,FDA 还可选择对临床试验用药品进行检查,如在信息不足以评估受试者风险或受试者将暴露于过度、显著风险的状况下”[3]。欧盟Directive 2001/20/EC指令人用药品临床试验质量管理规范中第十五条规定:“成员国应任命检查员对任何临床试验的相关场所进行检查,以验证对药物临床试验质量管理规范(Good Clinical Practice,GCP)和GMP的遵守情况,特别是试验场所、生产场所、用于分析的实验室等”。
目前我国无专门针对临床试验用药品制备过程的专项检查,药品上市前的药学研制和生产现场注册核查虽然会对制备过程有所涉及,但是核查一般从确证性临床试验用药品的制备开始,较少关注早期临床试验用药品的制备情况,且注册核查旨在核查真实性与一致性,而非临床试验用药品制备过程的GMP符合性[13-14]。因此,建议基于风险开展针对临床试验用药品制备过程的GMP符合性检查;另外,建议基于风险增加临床试验用药品抽样检验,这也可以在一定程度上促进临床试验用药品制备企业的质量管理水平提升,以促进附录的贯彻实施[15]。

来源:中国药事
关键词: 临床试验