
嘉峪检测网 2024-12-23 14:55
导读:统计美国食品和药物管理局(FDA)2019 年至 2023 年财政年度(简称财年)发布的药品项下的483 表格缺陷。
摘要:目的 提升企业药品生产质量管理水平。方法 统计美国食品和药物管理局(FDA)2019 年至 2023 年财政年度(简称财年)发布的药品项下的483 表格缺陷,分别按缺陷编号、引用条款号、药品生产质量管理规范(GMP)章节进行分析,并总结相应启示。结果综合缺陷编号及引用条款号,FDA 发现在质量管理、偏差管理、书面记录及工艺控制规程、实验室及微生物控制等方面的缺陷较多。结论企业应建立药品全生命周期的质量管理制度及处理偏差的操作规程,药品监管机构可利用信息化技术加强对药品的监管。
美国国会制定的The Federal Food,Drug,and Cos⁃metic Act FD & C Act(简称FD&C)及其他法律构建了美国食品和药物管理局(FDA)法律法规的基础,其中Code of Federal Regulations(CFR)第21 章211 部分(21 CFR & 211)为药品生产质量管理规范(GMP)[1]。FDA监管事务办公室(ORA)负责现场检查活动,以确认药品生产是否符合GMP要求。当检查员观察到可能构成违反FD & C和相关法案的情况时,会根据GMP相关标准形成483表格,即不符合GMP的缺陷清单列表。FDA检查员就483表格内容与药品生产企业管理层进行讨论,确保其充分理解检查缺陷,并在检查结束时将483表格发送给管理层。483表格代表FDA对是否违反FD & C法案或相关法规的最终决定,FDA会就483表格、检查报告、现场收集的所有证据或文件、企业回复整改情况及企业沟通情况综合形成最终的结论,并采取后续措施[2]。FDA透明度计划要求按财政年度(简称财年)公布检查缺陷项统计情况,该缺陷表涵盖食品、药品、兽药、医疗器械、生物制品等。本研究中统计了FDA 2019年至2023年财年发布的药品项下的483表格缺陷[3],分别按缺陷编号、引用条款号、GMP章节进行分析,并总结相应启示。现报道如下。
1、 检查总体情况
本研究中的483表格数量仅为FDA官方网站发布的数据,不包括发送给原料药生产商的483表格,以及电子系统外手动发布的483表格。2019年至2023年财年,FDA境内外共开展包括药品在内的所有检查57 495次,其中境内检查48 674次,境外检查8 821次[4]。详见图1。

受新冠肺炎疫情影响,2020年至2021年财年,检查次数大幅下降,其中2020 年财年境内外检查次数较2019年分别下降了48. 85%和59. 66%;2022年至2023年财年,检查次数稳步回升。受检查次数的影响,2019年至2023年财年分别发布了779份、349份、215份、466份、510份483表格[5]。
FDA综合483表格的回复发出检查结果[4],2009年至2019年财年,FDA平均每年发出的无需采取整改措施的结果(NAI)数量为13 626份,强制采取措施的结果(OAI)数量为706 份;2020 年NAI 数量降至6 033 份,OAI数量降至344份;2023年NAI数量升至12 628份,OAI数量升至439份,但尚未恢复到新冠肺炎疫情前的水平。
2、 以缺陷编号进行分析
2. 1 数据统计
FDA对21 CFR & 211中的条款进行拆分、细化、标准化引用,针对不同缺陷内容形成了唯一的内部缺陷编号。2019年至2023年财年,FDA分别引用了384个、286个、235个、283个、328个缺陷编号。引用数量排名前10的缺陷编号见表1。

2. 2 数据分析
在新冠肺炎疫情期间,FDA虽减少了检查频次,但引用频次较多的缺陷编号仍较稳定,同时检索2014年至2019 年财年FDA 的检查数据[6],其中缺陷编号1105,21 CFR 211. 22(d)[质量保证(QA)部门履职及规程书面化]连续10年排名第1,说明FDA对QA部门履职情况持续关注,且行业内对QA 部门履职情况距离FDA的要求还有差距。
排名前5 的缺陷编号较稳定,但缺陷编号1177,21 CFR 211. 63(设备设计、尺寸、布局)在2019年财年并未进入前10名,2020年、2022年、2023年财年均位居第5位,说明随着行业的不断发展,监管部门对设施设备的要求在不断提高,FDA对企业设备设计、尺寸、布局的关注度在不断增强。另外,缺陷编号3585,21 CFR 211. 110(a)(中间过程控制书面操作规程)及缺陷编号1434,21 CFR 211. 42(c)(10)(iv)(无菌产品的环境监测系统)2020 年至2022 年财年未进入前10 名,但在2023年财年分别位于第9名和第10名,说明FDA对中间过程控制及无菌药品的环境监测的重视程度在不断提高。
随着行业的不断发展,缺陷编号1215,21 CFR 211. 67(b)(设备清洁和维护的书面规程),缺陷编号1883,21 CFR 211. 165(a)(每批产品放行前需经实验室检测),缺陷编号1274,21 CFR 211. 68(a)(未按确保正常运行的书面规程进行自动/ 电子设备的常规校验检查),缺陷编号1112,21 CFR 211. 25(a)[人员培训(操作、GMP、标准操作规程< SOP >)],缺陷编号1809,21 CFR 211. 160(a)(遵循/ 记录实验室控制)均未进入2023年财年的前10名,说明以上几方面近几年进步较大;同时,2019 年财年排第10 名的缺陷编号1914,21 CFR 211. 166(a)(稳定性试验方案)在2020年至2023年财年均未进入前10名,说明企业在稳定性试验方案方面的操作已日渐规范。
3、 以引用条款号进行分析
3. 1 数据统计
FDA 211条款中,同一条款可对应多个子条款或多个缺陷编号,不同条款及不同缺陷编号对应不同情况,针对不同缺陷编号进行合并统计,2019年至2023年财年排名前10的引用条款见表2;根据不同缺陷编号及子条款进行合并,2023年财年排名前15的引用条款见表3。对比分析表1和表2引用条款发现,前4名缺陷条款编号基本一致,仅21 CFR 211. 192 及21 CFR 211. 22(d)顺序发生了改变。21 CFR 211. 110(a)(除2019年外)及21 CFR 211. 42(c)(10)(iv)均为新列入2023年财年引用排名前10的条款。以下针对引用条款排名前5及新列入2023年的条款进行分析。


3. 2 数据分析
条款21 CFR 211. 192(偏差审核、记录及调查):该条款强调在产品放行前,为保证确认与书面操作规程的符合情况,质量管理部门对所有药品生产与控制记录(包括包装与贴签)都应进行审核和批准。不管该批是否已销售,所有未经解释的偏离(包括超出主生产与控制记录中设定的最大或最小理论收率)或其任何原辅料不符合质量标准的情况均应进行彻底调查。对于同品种的其他批次及与偏差有关的批次均应进行调查,同时调查应有书面记录,并应包括调查结论及后续的纠正预防措施(CAPA)。在FDA历年的检查中,该条款缺陷主要集中在以下3个方面:1)未彻底调查出现偏差的情况,其中包括生产过程中及与质量标准不相符的情况;2)未对发生的偏差、偏差调查结论及后续采取的CAPA进行书面记录;3)发生的偏差未经QA进行审核。在FDA对德国某企业[7]的检查中,该企业的中间产品多次超过微生物符合行动限未进行偏差调查,并进行了产品放行,且该企业在2010年、2012年、2013年和2021年财年检查中重复发现此类缺陷,FDA对此发放了警告信。可见,FDA非常关注偏离正常生产规程及质量标准的情况,需要企业对发生偏差的原因进行彻底调查,同时采取适当的CAPA,以持续、不断地改进;同时,非常重视偏差的记录和审核,确保生产能持续、稳定地合规生产。
条款21 CFR 211. 22(d)(QA部门履职及规程书面化):FDA要求设置具有批准和拒绝所有原辅料、药品容器、密封件、中、间产品、包装材料、标签及药品的职责与权力的质量管理部门,且该部门有权审查生产记录,确保无差错,如出现差错,应确保展开充分调查。由FDA历年检查发现的缺陷可知,FDA不仅强调质量管理部门应制订书面管理规程,并对遵循其规程也同样重视。FDA发布的警告信[8]中提到,该企业的QA未对药品的生产进行充分监督,未履责的情形主要包括缺少对成品药品进行充分的微生物检验,未对不符合质量标准的情况进行充分调查,缺少专用生产设备的清洁和维护规程,未建立完善的稳定性计划及缺少产品质量回顾。FDA对QA部门在企业中是否拥有相对独立履行其职责的权利,质量授权人是否能免于企业负责人等其他人员干扰的独立性,QA部门能否高效开展工作,QA部门的所有职责是否都已编写并记录在案等非常关注,最终目标是确保企业拥有强有力的质量体系来维护并积极影响产品的质量。
条款21 CFR 211. 100(a)(书面记录及工艺控制规程):企业应制订生产和工艺控制的书面操作规程,以保证药品符合其质量标准,同时这些书面规程(包括任何变更)应由相应部门起草、审核和批准,并由质量管理部门进行审核和批准。FDA还强调人员的执行,书面规程能提供足够的信息,以确保人员可正确、一致地执行每项活动,且能形成恰当的文件记录,以便用于生产、QA和监管部门检查。在FDA发布的483表格中,该条款的问题主要集中在以下3个方面:1)缺少相应的书面规程;2)缺少QA对书面规程的审核和批准;3)书面规程的变更缺少QA的审核和批准。同时,在FDA发布的警告信中,该条款也多次涉及。在美国某公司的警告信[8]中,FDA认为其生产工艺缺少工艺验证;在另一家公司的警告信[9]中,该条款主要表明该公司未能提供支持性文件记录证明药品生产工艺经过验证,以及批生产记录中缺少关键工艺参数的记录。
条款21 CFR 211. 160(b)(实验室控制):实验室控制应包括制订科学可靠及恰当的质量标准、取样方案和检验方法,以保证原辅料、药品容器、密封件、中间产品、标签和药品符合适当的鉴别、规格、质量和纯度的标准。该条款同样在FDA的警告信中经常被提及,且情况多样。如企业未遵守质量标准进行检验,仅以无菌检查替代产品的质量评估[10];企业采取的灭菌前微生物负荷结果计算未灭菌成品批次的无菌保障水平的方法不科学[7],实验室不合格测试(OOS)结果调查不充分等。尽管FDA在2022年5月发布了行业指南Investigat⁃ing Out - of - Specification(OOS)Test Results for Phar⁃maceutical Production[11],从实验室人员职责、实验室调查阶段、可能需要的附加检测、扩大调查范围、检测结果的最终评估多维度对OOS结果调查进行了分析,但实验室控制仍需不断改进。
条款21 CFR 211. 113(b)(无菌药品微生物污染书面操作规程):应制订并遵循有关防止无菌药品微生物污染的书面操作规程,应包括所有无菌和灭菌工艺的验证。FDA发现的缺陷主要集中在以下2个方面:1)缺少相应规程的建立;2)防止无菌药品微生物污染的规程未经过验证。FDA在对印度一家企业[12]的检查中发现,企业的操作规程未要求在关键区的动态条件下进行烟雾试验,以及在某循环出料口未进行生物指示剂的评估以确保该位置的无菌环境。无菌和灭菌工艺的验证通常包括文件的建立,证明其工艺能稳定产出满足其预定用途和质量标准的产品。培养基灌装、操作控制、环境控制、产品无菌检验共同保证产品的无菌。
条款21 CFR 211. 110(a)(中间过程控制书面操作规程):应制订并遵循适当的中间过程控制及检验或检查的书面操作规程,以保证药品批次的均一性和完整性,同时应对中间控制规程进行监控,并对可能引起中间产品及药品特性变化的生产工艺程序进行验证。FDA发现的缺陷主要集中在以下3个方面:1)未建立监控中间过程控制的程序;2)缺少相应书面规程;3)过程控制程序有缺陷,如缺少混合均匀性及片剂或胶囊剂重量变化的检查。FDA在对美国某企业[13]的检查中发现,企业未对执行激光钻孔药品中间过程检查和可接受质量水平(AQL)进行检查,并缺少对操作人员的资质确认;企业未为激光钻孔药品的中间过程检查和AQL检查制订适当的书面规程;缺少对操作人员的视力、设施的照明和放大、操作人员佩戴的塑料面罩不会妨碍检查关键药品属性的能力的确认。
条款21 CFR 211. 42(c)(10)(iv)(无菌产品的环境监测系统):该条款要求操作应在明确规定且大小适宜的区域内进行,此条款中分为多个子条款,主要涉及待验产品的存放、不合格产品的存放、中间产品的贮存、包装贴签操作等区域的要求,对无菌工艺区域的要求主要涉及地板、温湿度控制、压强等方面,其中环境监测部分缺陷频次较高,主要是无菌加工区在环境条件监测系统方面存在缺陷。FDA在对加拿大一家企业[14]的检查中发现,企业所执行的环境监测频率过低,未覆盖所有生产区域,微生物监测的行动限设置不合理,缺少人员监测及设备与设施表面监测,监测水平不足以评估生产环境是否在受控状态下运行,企业关于环境微生物鉴定的信息过少。
4、 按章节进行分析
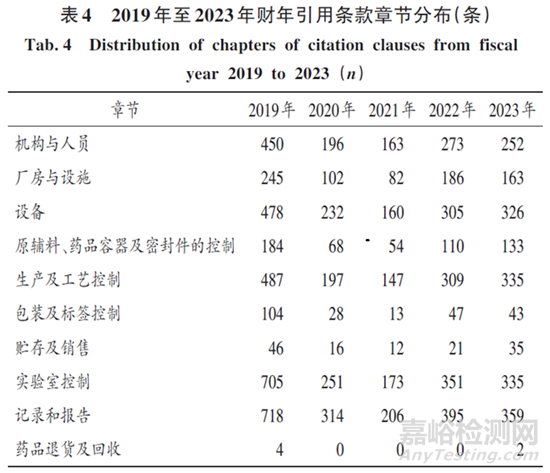
FDA的GMP共11个章节,按缺陷引用条款章节对2019年至2023年财年的数据进行分析发现,实验室控制、记录和报告2 个章节为引用条款数最多的章节。2023年的记录和报告、实验室控制、生产及工艺控制、设备、机构与人员条款的分布较均匀,厂房与设施,原辅料、药品容器及密封件的控制,包装及标签控制、贮存及销售的引用条款相对较少,药品退货及回收的引用条款最少。详见表4。
5、 启示
5. 1 药品全生命周期的质量管理
企业应保证QA部门的独立性,拥有能独立履行其职责的权利。在FDA检查缺陷分析中发现,缺陷基本指向其QA部门的履职职责,包括但不限于记录、审核等。因此,药品全生命周期中的质量管理尤为重要。产品生命周期管理始于研发,良好的研发质量管理体系能确保药品关键质量属性的识别与控制。国家药品监督管理局(NMPA)食品药品审核查验中心2021年发布的《药品注册核查要点与判定原则(药学研制和生产现场)》[15]要求,药品研制现场应建立与研究内容相适应的组织机构和质量管理体系。随着药品研发进入到临床试验用研究阶段,药品的制备不仅是实验室内的研究项目,且是将供研究用的临床样品作用于受试人群,管理要求随用药风险的增加而提高。2022年5月,NMPA发布了《< 药品生产质量管理规范(2010年修订)> 临床试验用药品附录》,对不同阶段药品的质量管理提出了相应的要求[16]。
随着上市许可人制度的发布,相关配套政策法规也不断出台。2023年10月,NMPA发布的《关于加强药品上市许可持有人委托生产监督管理工作的公告(2023年第132号)》中强调,“重点检查申请人关键岗位人员配备和在职在岗情况、质量管理体系建设和运行情况、对委托生产的管理情况等内容,确认申请人具备履行药品质量安全主体责任的能力”[17]。我国药品监管不断走向国际,NMPA发布公告,自2024年3月4日起,上市许可持有人开展的质量风险管理活动均适用国际人用药品注册技术协调会《Q9(R1):质量风险管理》指导原则[18],这对企业和监管部门均提出了更高的要求。
5. 2 偏差与OOS 调查
偏差是指对已批准的程序或规定标准的偏离[19],其中超出法定标准或企业制订的内控标准的数据为超标OOS结果。在药品生产过程中,偏离标准的情况不可避免,而偏差与OOS结果总是相伴发生,覆盖药品全生命周期,能识别偏差且认真对待至关重要。企业应建立处理偏差的操作规程,针对偏差的发现、报告、记录、调查、后续处理措施和CAPA,应采取基于风险的科学管理流程。同时,针对偏差与OOS调查的衔接,从实验室阶段进入全生命周期阶段的调查,应有清晰的认知。无论是FDA的检查,其他国际组织的检查,还是国内的注册核查或上市后检查,偏差及OOS调查均是检查的重点关注内容。我国对境外机构来华检查中也发现,偏差管理是重点发现缺陷的方面[20]。因此,建立良好的偏差管理制度,不仅是法规的要求,更是体现企业质量风险管理水平的重要手段与方法,也是保障药品质量的关键环节。
5. 3 标准化检查
FDA对21 CFR 211条款的细化、拆分非常细致,每种缺陷均对应不同的缺陷编号,同时定期更新。详细的分类情况可帮助政府监管部门掌握行业的发展情况,定期公布缺陷,能帮助企业不断对照并提高。FDA还不断探索新的标准化检查工具,在2016年就提出了新检查方案项目(NIPP),即使用标准化的电子检查方案,以更结构化的方式收集数据,从而实现更一致的设施监督和更快、更高效地分析。2022年,FDA年度报告提到NIPP可不断收集数据且使报告更标准化。随着信息化时代的到来,信息化技术能帮助监管机构更高效、精准地识别风险,利用大数据进行研判分析,让信息化技术为科学监管赋能。
参考文献
[1]张联. 美国药品检查机制对我国的启示[J]. 中国医药工业杂志,2021,52(7):971 - 974.
[2]FDA. FDA Form 483 Frequently Asked Questions[EB / OL].(2020 - 09 - 01)[2024 - 04 - 03]. https:// www. fda. gov /inspections - compliance - enforcement - and - criminal - in⁃vestigations / inspection - references / fda - form - 483 - fre⁃quently - asked - questions.
[3]姚立新,郑强. FDA 透明度计划回顾与展望[J]. 中国新药杂志,2018,27(3):255 - 266.
[4]FDA. Inspections,citations and published 483s data[EB / OL].(2020 - 09 - 01)[2024 - 04 - 03]. https:// datadashboard.fda. gov / ora / cd / inspections. htm.
[5]FDA. Inspection Observations[EB / OL].(2023 - 11 - 22)[2024 - 04 - 03]. https:// www. fda. gov / inspections - com⁃pliance - enforcement - and - criminal - investigations / in⁃spection - references / inspection - observations.
[6]葛渊源,张景辰,陈桂良. 美国FDA 2019 财年GMP 检查缺陷数据分析及启示[J]. 中国医药工业杂志,2020,51(2):276 - 283.
[7]FDA. Warning Letter Fresenius Kabi AG[EB / OL].(2023 -09 - 29)[2024 - 04 - 03]. https:// www. fda. gov / inspec⁃tions - compliance - enforcement - and - criminal - investiga⁃tions / warning - letters / fresenius - kabi - ag - 657085 -09142023.
[8]FDA. Warning Letter Colgin,Inc.[EB / OL].(2024 - 03 - 05)[2024 - 04 - 03]. https:// www. fda. gov / inspections - com⁃pliance - enforcement - and - criminal - investigations / warn⁃ing - letters / colgin - inc - 670332 - 01252024.
[9]FDA. Warning Letter Colonial Dames Company,Ltd.[EB / OL].(2023 - 12 - 26)[2024 - 04 - 03]. https:// www. fda. gov /inspections - compliance - enforcement - and - criminal - in⁃vestigations / warning - letters / colonial - dames - company -ltd - 659151 - 12052023.
[10]FDA. Warning Letter Signature Biologics,LLC[EB / OL].(2023 - 09 - 29)[2024 - 04 - 03]. https:// www. fda. gov /inspections - compliance - enforcement - and - criminal - in⁃vestigations / warning - letters / signature - biologics - llc -631039 - 09182023.
[11]FDA. Investigating Out - of - Specification(OOS)Test Re⁃sults for Pharmaceutical Production - Level 2 rev ⁃ision[EB / OL].(2022 - 05 - 16)[2024 - 04 - 03]. https://www. fda. gov / regulatory - information / search - fda - guid⁃
ance - documents / investigating - out - specification - oos -test-results-pharmaceutical-production-level-2-revision.
[12]FDA. Warning Letter Intas Pharmaceuticals Limited[EB / OL].(2023 - 11 - 28)[2024 - 04 - 03]. https:// www. fda. gov /inspections - compliance - enforcement - and - criminal - in⁃vestigations / warning - letters / intas - pharmaceuticals - lim⁃ited - 662868 - 11212023.
[13]FDA. Warning Letter Actavis Laboratories FL,Inc.[EB / OL].(2019 - 03 - 05)[2024 - 04 - 03]. https:// www. fda. gov /inspections - compliance - enforcement - and - criminal - in⁃vestigations / warning - letters / actavis - laboratories - fl -inc - 567857 - 02012019.
[14]FDA. Warning Letter Lernapharm(Loris) Inc.[EB / OL].(2018 - 09 - 11)[2024 - 04 - 03]. https:// www. fda. gov /inspections - compliance - enforcement - and - criminal - in⁃vestigations / warning - letters / lernapharm - loris - inc -552525 - 09042018.
[15]国家药品监督管理局食品药品审核查验中心. 国家药品监督管理局食品药品审核查验中心关于发布《药品注册核查工作程序(实行)》等5 个文件的通告》(2021 年第30 号)[A / OL].(2021 - 12 - 20)[2024 - 04 - 03]. https:// www. cfdi. org.cn / resource / news / 14199. html.
[16]国家药品监督管理局. 国家药监局关于发布《药品生产质量管理规范(2010 年修订)》临床试验用药品附录的公告(2022年第43号)[A/OL].(2022-05-27)[2024-04-03].
https:// www. nm pa. gov. cn / xxgk / fgwj / xzhgfxwj /20220527182006196. html.
[17]国家药品监督管理局. 国家药监局关于加强药品上市许可持有人委托生产监督管理工作的公告(2023 年第132号)[A / OL].(2023 - 10 - 23)[2024 - 04 - 03]. https://
www. nmpa. gov. cn / xxgk / fgwj / xzhgfxwj / 20231023160426145. html.
[18]国家药品监督管理局. 国家药监局关于适用《Q9(R1):质量风险管理》国际人用药品注册技术协调会指导原则的公告(2023 年第114 号)[A / OL].(2023 - 09 - 05)[2024 -04 - 03]. https:// www. nmpa. gov. cn / xxgk / ggtg / ypggtg /ypqtggtg / 20230905172504180. html.
[19]ICH. Q7:Good Manufacturing Practice Guide for Active Phar⁃maceutical Ingredients[EB / OL].(2000 - 11 - 10)[2024 -04 - 03]. https:// www. cde. org. cn / ichWeb / guideIch /downloadAtt / 1 / ba5110faef2acef5072ec3e58ab22cc0.
[20]徐长波,翟铁伟. 我国化学药生产企业境外检查观察情况分析[J]. 中国药业,2024,33(3):38 - 42.
内容来源:药事管理 2024 年12 月5 日第33 卷第23 期

来源:Internet
关键词: GMP检查