
嘉峪检测网 2025-04-19 10:42
导读:今天我们写在制药机械/设备计算机化系统验证这个流程中,有些什么详细的内容文件以及谁会提供这部分的文件,即流程与分工的问题。
今天我们写在制药机械/设备计算机化系统验证这个流程中,有些什么详细的内容文件以及谁会提供这部分的文件,即流程与分工的问题。我们的关注点不应该放在工具本身上,而在于关注这个工具能够为我们做什么,为我们带来什么结果。总之,该谁做什么,谁擅长做什么,谁就该做什么,不要一揽子由用户来做,也不需要事后完全依靠第三方来做,很简单的事情,谁该做的谁来做就可以了。
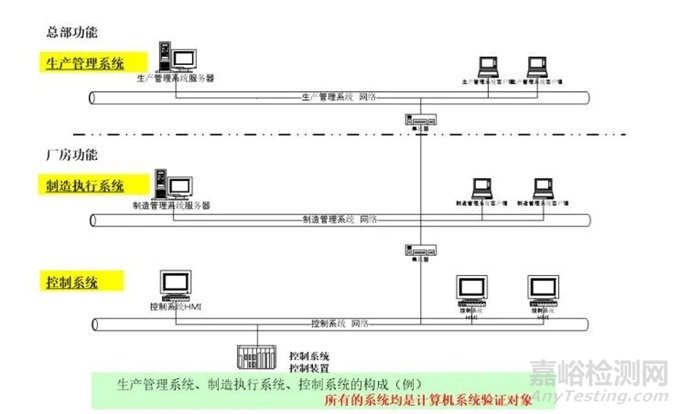
制药机械/设备计算机系统验证(CSV,Computer System Validation) 是确保制药设备及其相关计算机系统符合GMP(药品生产质量管理规范)和其他法规要求的关键过程。CSV的目标是证明计算机系统能够 consistently(一致地)满足其预定用途,并确保数据的完整性、可靠性和安全性。
计算机化系统的验证主计划可以作为《*****企业验证主计划》或者《***项目验证主计划》的一部分,说明计算机化系统的硬件、软件的分类原则、风险管理原则、确认与验证原则、确认与验证的控制策略,规定企业如何开展计算机化系统的验证工作。也可以单独制定《计算机化系统的验证主计划》,不过计算机系统通常作为设备的不可分割的一部分(管理类计算机系统除外),或者作为电气自控设备的一种,是否单独对待也是仁者见仁,智者见智的。
1. 验证流程
CSV通常遵循 V模型(V-Model)或 生命周期模型,分为以下几个阶段:
1.1 用户需求说明(URS,User Requirement Specification)
内容:定义系统的功能需求、性能要求、法规要求(如GMP、21 CFR Part 11)和用户期望。
输出:URS文档。
责任方:用户部门(如生产、质量控制)、IT部门、供应商。
1.2 功能需求说明(FS,Functional Specification)
内容:基于URS,详细描述系统的功能、操作流程和技术要求。
输出:FS文档。
责任方:供应商、系统工程师。
1.3 系统设计说明(DS,Design Specification)
内容:描述系统的技术设计,包括硬件、软件架构、数据流和接口。
输出:DS文档。
责任方:供应商、系统工程师。
1.4 配置与开发
内容:根据DS进行系统配置和软件开发。
输出:配置好的系统或软件。
责任方:供应商、开发团队。
1.5 安装确认(IQ,Installation Qualification)
内容:确认系统已正确安装,硬件和软件符合设计规范。
输出:IQ报告。
责任方:验证团队、供应商、IT部门。
1.6 操作确认(OQ,Operational Qualification)
内容:测试系统的功能是否满足FS要求,包括正常操作、边界条件和异常情况。
输出:OQ报告。
责任方:验证团队、用户部门。
1.7 性能确认(PQ,Performance Qualification)
内容:在实际生产环境中测试系统,确认其能够 consistently(一致地)满足URS要求。
输出:PQ报告。
责任方:验证团队、用户部门。
1.8 验证总结报告(VSR,Validation Summary Report)
内容:总结整个验证过程,确认系统已验证并可以投入使用。
输出:VSR文档。
责任方:验证团队、质量保证(QA)部门。
1.9 变更控制与再验证
内容:对系统的任何变更进行评估,必要时进行再验证。
输出:变更控制记录、再验证报告。
责任方:变更控制委员会、验证团队。
2. 分工与责任
CSV是一个跨部门协作的过程,涉及多个团队和角色:
2.1 用户部门(生产、质量控制)
负责定义用户需求(URS)。参与测试(OQ、PQ)并提供反馈。确认系统是否满足实际生产需求。
2.2 IT部门
负责系统的安装、配置和维护。确保系统的数据完整性和安全性。支持验证团队进行IQ和OQ测试。
2.3 供应商
提供符合URS和FS的系统或软件。参与系统设计、开发和配置。支持验证团队进行IQ、OQ和PQ测试。
2.4 验证团队
制定验证计划(Validation Plan)。执行IQ、OQ、PQ测试并编写报告。协调各部门的验证活动。
2.5 质量保证(QA)部门
审核和批准验证文件(URS、FS、IQ、OQ、PQ、VSR)。确保验证过程符合GMP和其他法规要求。监督变更控制和再验证。
2.6 项目管理团队
负责验证项目的整体进度和资源分配。协调各部门的沟通与协作。
3. 关键考虑因素
数据完整性:确保系统符合ALCOA+原则(可追溯、清晰、同步、原始、准确、完整、一致、持久、可用)。
法规合规性:确保系统符合GMP、21 CFR Part 11(电子记录和电子签名)、EU Annex 11等法规要求。
风险管理:在验证过程中识别和评估风险,并采取适当的控制措施。
文档管理:所有验证文档必须完整、准确并妥善保存。
4. 常见挑战
需求不明确:URS定义不清可能导致后续验证失败。
变更管理:系统变更未及时记录或评估可能导致验证状态失效。
资源不足:验证过程需要大量时间和人力,资源不足可能影响进度。
供应商支持不足:供应商未能提供足够的技术支持可能延误验证。

来源:Internet