
嘉峪检测网 2025-05-08 08:24
导读:本文介绍了反义寡核苷酸(ASOs)药物的吸收、分布、代谢和排泄特征。
反义寡核苷酸(ASOs)被认为是目前最先进的RNA类治疗手段之一。ASOs是长度为12–25个核苷酸的互补核酸片段,被设计为特异性地与内源性前mRNA或mRNA的互补序列杂交,以调节生物功能。它们通过以下方式实现这一目标:通过酶促裂解降解mRNA,改变前mRNA剪接以包含或排除特定内含子/外显子,或者改变调控RNA的功能。其他潜在的ASOs作用机制还包括通过阻碍核糖体活性诱导翻译停滞、通过抑制剪接干扰前mRNA成熟、在细胞核内使前mRNA不稳定,或者纠正隐性剪接位点。ASOs的序列专门针对疾病病理学背后的遗传异常。迄今为止,美国FDA已批准10种ASO药物用于治疗遗传性疾病,例如杜氏肌营养不良症(DMD)、脊髓性肌萎缩症(SMA)和家族性淀粉样多发性神经病。
化学修饰的ASOs设计
Stephenson和Zamecnik首次将ASOs用于治疗目的,并证明了基于DNA的ASOs可以在体外抑制病毒复制。然而,由于核酸酶对磷酸二酯骨架的降解以及蛋白质结合能力差,导致其无法在体内有效分布到组织中,这些发现未能在体内得到持续验证。因此,大量的研究工作都集中在对ASO进行化学修饰,以提高其稳定性、组织靶向性、摄取能力、药代动力学和药效学特性。
对磷酸二酯骨架的修饰产生了新的化学结构,例如磷酰胺、硫代磷酸酯、硼烷磷酸酯、甲基磷酸酯和磷酸酯类似物,而核糖单元则被修饰为2'-O-甲基、2'-氟或4'-硫代核糖,或者被替换为吗啉环。此外,还评估了用非天然或修饰的碱基(如次黄嘌呤、7-去氮鸟嘌呤、2,6-二氨基嘌呤、4-硫代尿嘧啶和二氟甲苯)替换DNA/RNA核苷酸碱基,以提高代谢稳定性和增强杂交亲和力。目前具有临床应用价值的两大ASO平台是磷酰胺吗啉寡核苷酸(PMOs)和硫代磷酸酯(PSs)。
ASOs ADME特征
吸收和分布
ASO药物的吸收取决于其不同的理化特性,包括离子化、酸解离常数(pKa)、疏水性以及给药途径。药物的分布则取决于给药途径、游离药物浓度、组织血流灌注、组织结合、局部pH以及细胞膜的通透性。
第一代和第二代PS修饰寡核苷酸由于其骨架带负电荷,能够与血浆蛋白(特别是白蛋白)高度结合(结合率> 85%)。例如,Mipomersen在人体和小鼠中的蛋白结合率分别为95%、85%。而基于PMO修饰的ASOs是不带电的,在人体中的蛋白结合率较低(6%-17%),从而限制了其组织吸收。例如,PMO类药物Eteplirsen人血浆蛋白结合率为6.1%-16.5%。由于血浆蛋白结合与细胞摄取和肾小球滤过相关,PS-ASOs与PMOs相比具有更持久的组织分布和更慢的尿液排泄特性,这可以通过它们的蛋白结合差异来解释。
口服给药的PS-ASOs和PMOs由于分子量较大且亲水性强,导致其膜通透性低,吸收效果差。虽然口服给药不可行,但通过化学修饰可以实现静脉或皮下注射后的系统暴露。目前,大多数ASOs通过静脉注射给药,以确保生物利用度最高。这种给药方式使得药物能够快速分布至血管化程度高的器官(如肝脏、肾脏和脾脏)。不过,ASOs分布到心脏、肌肉和肺等组织的速度较慢,因此需要更频繁的给药。比如,临床试验中,每周一次静脉注射Eteplirsen(30 mg/kg),其平均表观分布容积(Vd)为601 ml/kg,提示组织中的分布较高。单次或多次静脉注射后的Cmax通常出现在注射结束时,且多次给药PK呈剂量比例关系。
另外,由于寡核苷酸药物不具有膜通透性,它们通过吞噬作用或受体介导的内吞作用而不是被动扩散进入细胞。细胞摄取过程大致可以分为两个步骤:ASOs吸附到细胞表面蛋白和内化。许多细胞表面受体参与ASOs的受体介导内吞作用,包括表皮生长因子受体、G蛋白偶联受体和清道夫受体。特别是关于清道夫受体的研究有很多,发现它们介导裸ASOs的摄取,例如stabilin-1和stabilin-2介导PS-ASOs的肝脏摄取,清道夫受体A1介导PMOs的肌肉摄取。内化的ASOs需要从内体中释放出来,才能与细胞质或细胞核中的目标RNA相互作用。这些细胞内ASO的运输过程被认为受到细胞质和细胞核区域中多种蛋白的相互作用调控。通过使用生物素标记的PS-ASOs进行亲和力选择,Liang等人鉴定了一组与PS-ASOs相互作用的细胞内蛋白。最近的一项研究表明,在早期内体途径中,Rab5C和早期内体抗原1(EEA1)参与了PS-ASOs的运输,并在stabilin介导的内化后促进它们的内体逃逸;在晚期内体途径中,Rab7A和溶酶体双磷酸甘油磷脂参与了PS-ASOs的运输。与PS-ASOs相比,关于PMOs细胞内运输机制的分子基础研究较少。
为了进一步优化ASOs的组织分布情况,目前正在研究将ASOs与脂肪酸或肽结合。比如(GalNAc3)ASO结合物已经进入临床阶段,本品通过唾液酸糖蛋白受体被肝细胞主动摄取,显著增强了对肝细胞的靶向递送,从而改善了药效。
另一种成功提高ASOs穿透能力的方法是通过与细胞穿透肽(CPPs)结合。肽的正电荷有助于与不带电的PMO ASO结合。研究表明,CPPs通过内吞机制促进PMO进入细胞,该机制涉及CPP的阳离子与细胞表面蛋白多糖的阴离子之间的相互作用。CPPs已被用于提高PMOs在肌肉组织中的细胞摄取。
另外,如前文所述,给药途径对ASO产品的分布有影响。与IV相比,SC给药后的ASO分布半衰期通常更长,可能是由于药物从注射部位逐渐吸收所致。一项在非人灵长类动物中进行的研究显示,通过IV或SC给予320 mg/kg的Eteplirsen,显示SC给药后的生物利用度为100%,并且Cmax出现在大约药后7小时。Mipomersen在猴子中也显示出可以从皮下注射部位完全吸收,生物利用度为100%,Cmax出现在3-4小时。
当然,对于IV或SC给药无法到达的靶部位,直接局部ASO给药也是可行的。局部给药可用于眼部、中枢神经系统等系统给药难以到达的靶组织。以Fomivirsen为例,通过玻璃体注射将药物局部递送至眼睛,只需要少量药物,并且可以直接分布到视网膜。鞘内给药可以绕过血脑屏障,使ASO能够递送至中枢神经系统。尽管具有一定的侵入性,但鞘内给药已被证明在增加ASO在大脑和脊髓中的生物利用度方面是有效的,并且与IV和SC相比,减少了全身暴露。Nusinersen的鞘内给药展示了ASOs在CNS领域应用的潜力,并为靶向新的适应症(如亨廷顿病和肌萎缩侧索硬化症)打开了大门。成年食蟹猴中进行的单次鞘内注射结果显示,药物在脊髓中广泛分布,并达到具有药理活性的水平。此外,给药后7天的组织分析结果显示,nusinersen在CNS组织中的总量呈剂量依赖性,且在脊髓和皮层中的分布更多。即使在最低剂量(1 mg/kg)时,目标组织中的药物水平也比EC50(~1μg/g)高出3-8倍。
鼻内给药正在探索作为ASO用于CNS适应症的给药方式之一。鼻内给药后,ASO分子可以通过嗅神经和三叉神经通路等进行转运。使用CPP可以进一步增强CNS的递送,且已经在siRNA中取得成功。
因胃肠道吸收有限,口服给药对ASOs是一种挑战。PS类ASO ISIS 14725在大鼠中通过十二指肠给药后的生物利用度约为5.5%,而PMO类AVI-4472的口服生物利用度在大鼠中为41%-79%。然而,尚未见PMOs在人类中的口服给药情况的报道。
代谢
虽然ASOs主要被核酸酶代谢,但其代谢稳定性因不同的平台和种属而有所差异,主要取决于其化学骨架(如下表所示)。

核酸酶分为外切酶和内切酶,外切酶主要切割ASO的末端位点,识别3′-羟基。内切酶则切割ASO的内部部分,识别核糖上的2′-羟基。缺乏3′和5′末端核糖修饰的ASOs主要被外切酶降解,生成3′或5′缩短的片段,这些片段可能仍具有反义活性,但也会被组织中的内切酶进一步降解。
Defibrotide是一种具有天然磷酸二酯骨架的ASO混合物,它被血浆外切酶和多种DNA降解酶迅速代谢,释放出游离的2′-脱氧核糖、嘌呤和嘧啶碱基。因此,Defibrotide在2小时静脉输注后的半衰期仅为14分钟。
再看下PS修饰ASOs的代谢。Mipomersen和Nusinersen是第二代PS修饰ASOs,其代谢主要发生在硫代磷酸酯骨架上,通过核酸酶的作用。Mipomersen代谢由内切酶和外切酶介导,形成不再具有药理活性的短序列代谢产物。Nusinersen通过鞘内给药递送至大脑后,其代谢由外切酶3′和5′介导的水解作用完成,切割末端核苷酸,产生缩短的ASO代谢产物。由于大脑的代谢活性较低,nusinersen在给药后1年仍可在大脑中被检测到,反映了其在脑脊液中的长半衰期(135-177天)。Fomivirsen是通过玻璃体注射给药的PS修饰ASO。Fomivirsen在人体中的清除速度较慢,半衰期约为55小时。临床前研究表明,分布到视网膜的Fomivirsen通过外切酶消化缓慢代谢。另外,对于PS-ASOs,已观察到小鼠、猴子、大鼠和人类的代谢途径相似,表明PS-ASOs的代谢途径在种属间没有显著差异,且PS-ASO代谢物的系统暴露量通常较低。
PMOs修饰的ASOs通常在核酸酶下非常稳定。如Eteplirsen与血浆和肝细胞组分共孵育时,未形成任何代谢产物。然而,对于PPMOs(肽偶联PMO),其CPP序列可能会引入一个代谢位点,容易被广泛存在的蛋白酶降解。
其他偶联ASOs的代谢情况。例如,3H-ION-681257是一种基于PS的ASO,与GalNAc3偶联,给予大鼠后观察到大量肝脏分布,主要代谢途径是氧化,形成14个与GalNAc3相关的连接子代谢产物和游离ASO。胆汁排泄的代谢产物与肾脏排泄的代谢产物相同,除了氧化的线性和环状连接子代谢产物。3H-ION-681257被N-乙酰-β-葡萄糖苷酶、脱氧核糖核酸酶II、碱性磷酸酶和醇/醛脱氢酶代谢。
ASOs不是CYP450酶的底物,尚未报告任何已批准的ASOs存在CYP介导的代谢产物。此外,由于它们对CYP酶的亲和力低,ASOs尚未显示出与其他小分子药物存在药物相互作用(DDIs)。几项临床试验旨在评估ASOs与华法林(CYP2C9/3A4)、辛伐他汀(CYP3A4)、依折麦布(多种尿苷二磷酸葡萄糖醛酸转移酶)、罗格列酮(CYP2C8/2C9)、格列吡嗪(CYP2C8/2C9)、二甲双胍(肾脏)、顺铂(肾脏)和吉西他滨(核苷激酶)的潜在药物相互作用,结果显示不存在药物相互作用风险。最近一项研究考察了ISIS 304801、ISIS 420915、ISIS 681257和ISIS 396442,结果表明这些受试ASOs均未显示出对CYP1A2、CYP2B6、CYP2C8、CYP2C9、CYP2C19、CYP2D6、CYP2E1或CYP3A4的潜在抑制作用。此外,在冻存的人类原代肝细胞中,这些受试化合物在酶活性水平或mRNA水平上均未显示出对CYP1A2、CYP2B6或CYP3A4的诱导作用。这些数据与之前报道的ASOs与CYP酶之间无相互作用的结果一致。
排泄
ASOs主要以原型药通过尿液或粪便排泄。然而,尿液或粪便中排泄的程度取决于ASOs的化学性质、给药途径、动物种属和偶联情况。蛋白质结合的程度也会影响排泄,因为与血浆蛋白的结合使ASOs免于肾小球滤过,从而减缓肾脏清除(ASOs清除的主要途径)。PMO修饰ASOs的高肾脏清除率可能与低蛋白结合率相关,导致其较短的血浆半衰期。
在肾功能正常的健康志愿者中,大约有10%-14%的Defibrotide以原型形式通过尿液排泄,19%通过粪便排泄。然而,在肾功能受损的患者中,AUC增加了50%-60%。肾功能受损的患者需要特别监测,并进行必要的剂量调整。
蛋白质结合在排泄中的作用也体现在mipomersen上,这是一种第二代PS修饰的ASO。带负电荷的2′修饰PS与白蛋白结合,使其免于通过肾小球滤过被清除。小鼠25 mg/kg皮下注射后,24小时内有23%的mipomersen通过尿液排泄,而5 mg/kg皮下注射后这一比例仅为1%。Mipomersen在小鼠体内的清除率高于人类,小鼠的血浆清除率为674 ml/h/kg,半衰期为0.33小时,而人血浆清除率为40.9 ml/h/kg,半衰期为1.26小时,可能与小鼠的蛋白结合率较低有关。
3H-ION-681257是一种第二代PS ASO,与THA-C6-triantennary GalNAc3偶联,通过唾液酸糖蛋白受体实现肝细胞的靶向递送。由于在肝脏中的特异性分布,3H-ION-681257被迅速代谢并排泄,在给药后48小时内,尿液和粪便中分别回收了26%和71%的放射性。尽管肾脏清除是大多数ASOs的主要排泄途径,但nusinersen(第二代PS ASO)的肾脏排泄并不明显。在第三次给药后的第85天,尿液中检测到约0.5%的原型药物;不过,由于未追踪其代谢产物,不能排除肾脏是主要排泄途径的可能性。这种低肾脏排泄很可能是因为其在组织中的长半衰期(在猴子的脊髓和大脑中超过100天)以及从中枢神经系统组织到全身循环的缓慢清除过程。
临床试验表明,Eteplirsen PMO主要以原型药形式通过尿液排泄,经50 mg/kg静脉输注后,69.4%被排泄。Eteplirsen在50 mg/kg静脉输注后的总清除率为5.3 ml/min/kg,肾脏清除率在相同剂量下为3.9 ml/min/kg,占总系统清除率的三分之二。
AVI-6002和AVI-6003是以前分别用于埃博拉病毒和马尔堡病毒暴露后预防的联合疗法。AVI-6002由AVI-7537和AVI-7539组成,AVI-6003由AVI-7287和AVI-7288组成。这四种成分都属于PMOplus平台,该平台在骨架上含有特定位置的正分子电荷。在临床试验中,当这四种成分被给予健康志愿者时,PMOplus药物都在给药后的前24小时内通过尿液排泄。这四种药物观察到了剂量依赖性的肾脏清除:AVI-7537、AVI-7539、AVI-7287、AVI-7288分别有44%、31%、39%、52%以原型药形式被排泄。
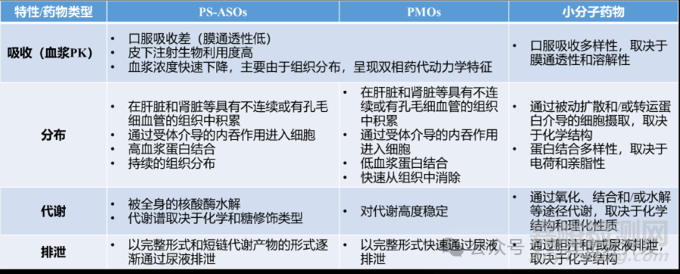
文章最后,简单总结下ASOs与传统小分子的DMPK异同,如下表所示。

来源:药理毒理开发