
嘉峪检测网 2025-05-12 19:38
导读:近日,ECA 分析质量控制组(AQCG)制定了一份新文件:《实验室数据管理指南——取样与样品管理》。
近日,ECA 分析质量控制组(AQCG)制定了一份新文件:《实验室数据管理指南——取样与样品管理》。 该文件强调了取样作为一个潜在的误差产生过程的至关重要性,并详细说明了制定可靠取样方案的要求,以将变异性降至最低并保持数据完整性。此外,该指南阐述了取样过程中涉及的关键步骤,从统计取样计划到取样方案设计,以及取样记录文件的编制等。
文件提及:取样过程中,物料包装密封容器的开启、取样及重新密封方式应能防止其内容物受到污染,同时防止对其他成分、药品容器或密封件造成污染。
文件提及:如果需要从容器的顶部、中部和底部对组件进行取样,则不得将此类分段取样混合用于检测。
文件提及:在物料质量被判定为合格之前,不得投入使用;在产品质量被判定为合格之前,不得进行销售或供应。
文件还介绍了WHO 针对物料的三种简化抽样方案:
n方案(即根号N+1方案):WHO强烈警示了使用该方案的情况。对于那些需要对所接收的用于生产药品的起始物料进行分析并决定放行或拒收的生产企业的质量控制实验室而言,不建议使用n方案。
p 方案:当物料性质均一,且来自已认可的来源,同时主要目的是进行鉴别检验时,可以使用“p方案”
r 方案:当物料被怀疑性质不均一,以及/或者物料来自一个不太知名的来源时,可以使用 “r 方案”
该文件还提供了实际示例和说明性图表,以帮助实验室实施有效的取样策略并确保符合法规要求。
文件翻译如下:
LaboratoryDataManagementGuidance –Sampling
andSampleManagement–
实验室数据管理指南——取样与样品管理
目录
1 Introduction to AQCG & its Guidances
分析质量控制小组(AQCG)及其指南简介
1.1 Guides and Guidances
指南和指导意见
2 Importance of Sampling and Sample Management
取样和样品管理的重要性
2.1 Sampling
取样
2.2 Sample Management
样品管理
3 Regulatory Requirements for Sampling and Sample Management
取样及样品管理的监管要求
3.1 USA cGMP (21 CFR §210ii & 21 CFR §211iii)
3.2 EU GMP
3.3 United States Pharmacopeia
美国药典
3.4 WHO Guidance for sampling of pharmaceutical products and related materials
WHO 药品及相关物料的取样指导意见
3.5 FDA Guidance Investigating Out-of-Specification (OOS) Test Results for Pharmaceutical Production, May 2022
2022 年 5 月 FDA 药品生产中OOS调查指南
3.6 GOOD Samples: Guidance on Obtaining Defensible Samples
良好取样:获取可靠样品的指南
4 Theory of Sampling
抽样理论
5 Nomenclature of sampling
抽样的术语
6 The Sampling Process
抽样过程
6.1 Statistical Sampling Plans
统计抽样计划
6.2 Acceptance Sampling Plans
验收抽样计划
6.3 WHO guidelines; three reduced sampling plans for starting materials
WHO 针对起始物料的三种简化抽样方案
7 Sampling Procedure & Protocol
取样程序&方案
7.1 In-process sampling
过程中取样
8 Sampling Record
取样记录
9 Additional Guidance sources on sampling
关于取样的其他指南
10 Sampling is only part of the Analysis and Testing Process
取样只是分析和测试过程的一部分
11 References
参考文献
1 Introduction to AQCG & its Guidances
分析质量控制小组(AQCG)及其指南简介
The AQCG aims at providing a networking platform for Analytical Chemists and Scientists and to facilitate an active discussion of the latest regulatory requirements as well as identifying and addressing technical issues and challenges. In addition, it supports a harmonised approach through providing discussion/position papers and generic guidances via expert working groups to foster an effective and efficient communication between industry, competent authorities and the pharmacopoeias.
分析质量控制小组(AQCG)旨在为分析化学家和科学家提供一个交流平台,推动对最新法规要求的积极讨论,同时识别和解决技术问题与挑战。此外,该小组通过专家工作组提供讨论文件和立场文件以及通用指南,支持采用统一的方法,以促进行业、主管部门和药典之间进行有效且高效的沟通。
1.1 Guides and Guidances
指南和指导意见
This guidance continues the sequence of these already published guidances which are available as pdf files for members of ECA.
本指南延续了此前已发布的一系列指导意见,这些指导意见以 PDF 文件的形式提供给欧洲化学工业协会(ECA)的会员。
• ECA _AQCG_ SOP 01_OOS_v2.0 August 2012
• ECA _AQCWG_ SOP 02_OOE OOT_v1.2_July 2023
• ECA _AQCG_ SOP 03_APLM_v1.0_July 2018
• ECA-AQCG-AIQSV-Guide-November 2023-v1.0
• ECA_Data Integrity Guide_v3.0 December 2022
2 Importance of Sampling and Sample Management
取样和样品管理的重要性
It is important to understand that sampling is always an error generating process and that although the reported result may be dependent upon the analytical method, it will always be dependent upon the sampling process.
取样往往是一个会产生误差的过程,而且尽管所报告的结果可能取决于分析方法,但它往往也取决于取样过程。
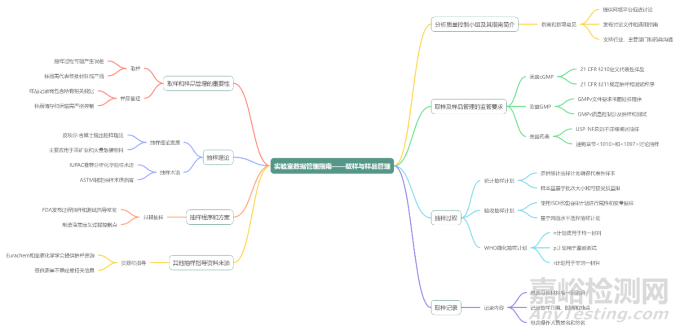
The sampling process includes the specific activities undertaken to select the sample from a batch or lot and the management of the chain of custody of that sample throughout the lifecycle journey from sampling, through testing to retention and ultimate disposal. A high level process flow is shown in Figure 1.
取样过程包括从批次中选取样品所进行的特定活动,以及从取样、测试到留样和最终处置的整个生命周期中对该样品的流转的管理,其高层次的流程如图 1 所示。
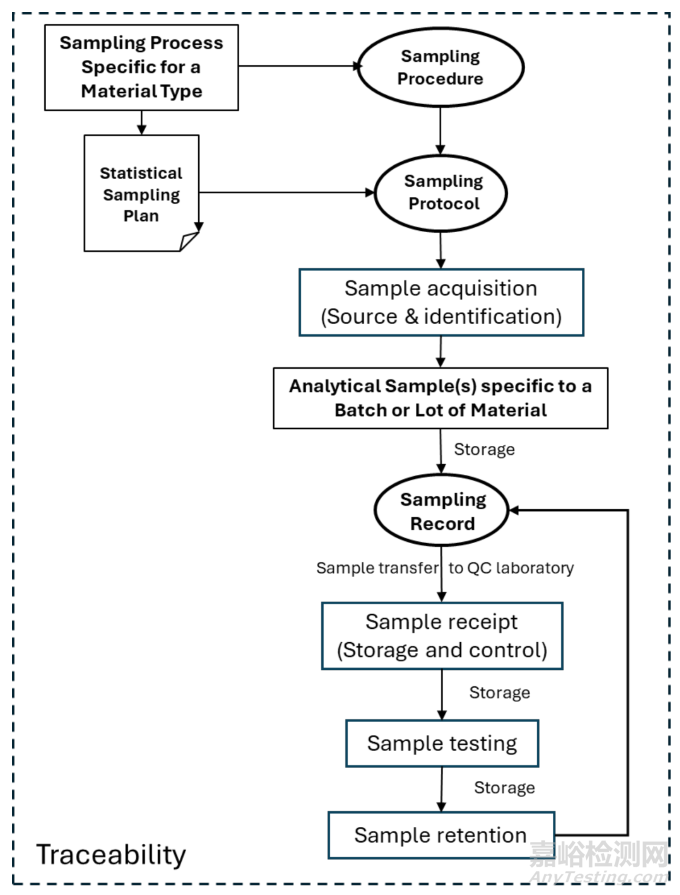
Figure 1 High level sampling and sample management process flow
图 1 高层次的取样及样品管理流程
2.1 Sampling
取样
Inconsistent sampling processes can lead to errors, resulting in data integrity issues and potentially incorrect decisions about pharmaceutical quality.
取样过程不一致会导致误差,进而引发数据完整性方面的问题,还可能使有关药品质量的决策出现错误。
If the sample provenance cannot be assured such that the sample is representative of the overall batch or lot of material or product any testing is useless. It is essential that sampling process steps and flows are consistent and traceable
如果无法保证样品的来源,使得样品不能代表整批物料或产品,那么任何检测都是徒劳的。至关重要的是,取样的流程步骤必须一致且具有可追溯性。
2.2 Sample Management
样品管理
Traceability is critical to demonstrate that the sample, as taken in accordance with the procedure is identified, protected and stored correctly prior to analysis. The sample record needs to contain all relevant data to confirm assurance that the sample tested has not been compromised prior to analysis in any way.
可追溯性至关重要,它能够证明按照程序采集的样品得到了识别、保护,并且在分析之前得到了正确的储存。样品记录需要包含所有相关数据,以确保所测试的样品在分析前未以任何方式受到影响。
In particular, the storage and transportation of samples activities should be strictly controlled.
特别是,样品的储存和运输活动应当受到严格控制。
Ideally sampling should be performed in an area or booth designed for and dedicated to this purpose. However, if this is not possible sampling facilities should be designed to
理想情况下,取样应在专门为此目的设计的区域或层流罩内进行。然而,如果无法做到这一点,取样设施的设计应……
prevent contamination of the opened container, the materials and the operator;
防止已打开的容器、物料以及操作人员受到污染;
prevent cross-contamination by other materials, products and the environment;
防止受到其他物料、产品以及环境的交叉污染;
protect the individual who samples (sampler) during the sampling procedure.
在取样过程中保护进行取样人员
3 Regulatory Requirements for Sampling and Sample Management
取样及样品管理的监管要求
The requirements for sampling and sample management are scattered across multiple sources as illustrated in Figure 2. Details are provided in the subsequent sections 3.1 to 3.6 and their subsections.
取样和样品管理的要求分散在多个来源中,如图2 所示。具体细节在后续的 3.1 至 3.6 节及其子节中给出。
Figure 2 Overview of sources of requirements for sampling and sample management
图 2 取样和样品管理要求的来源
3.1 USA cGMP (21 CFR §210ii & 21 CFR §211iii)
3.1.1 21CFR§210.3 Definitions
Representative sample means a sample that consists of a number of units that are drawn based on rational criteria such as random sampling and intended to assure that the sample accurately portrays the material being sampled.
代表性样品是指根据随机取样等合理标准抽取的由若干个单元组成的样本,旨在确保样本能够准确地描绘被取样的物料。
3.1.2 21 CFR §211.84 Testing and approval or rejection of components, drug product containers, and closures.
成分、药品容器和密封件的检测、批准或拒收
(a) Representative samples of each shipment of each lot shall be collected for testing or examination. The number of containers to be sampled, and the amount of material to be taken from each container, shall be based upon appropriate criteria such as statistical criteria for component variability, confidence levels, and degree of precision desired, the past quality history of the supplier, and the quantity needed for analysis and reserve where required by § 211.170.
每批货物的每个批次都应采集代表性样品进行测试或检验。待取样的容器数量以及从每个容器中抽取的物料数量,应基于适当的标准来确定,如成分变异性的统计标准、置信水平、所需的精确程度、供应商过去的质量记录,以及需要时,§211.170 所要求的分析和留样的数量。
(b) Samples shall be collected in accordance with the following procedures:
样品应按照以下程序采集:
1) The containers of components selected shall be cleaned when necessary in a manner to prevent introduction of contaminants into the component.
所选定的成分容器如有必要,应进行清洁,其清洁方式应能防止污染物进入成分中。
2) The containers shall be opened, sampled, and resealed in a manner designed to
prevent contamination of their contents and contamination of other components,
drug product containers, or closures.
容器的开启、取样及重新密封方式应能防止其内容物受到污染,同时防止对其他成分、药品容器或密封件造成污染。
3) Sterile equipment and aseptic sampling techniques shall be used when necessary.
必要时,应使用无菌设备并采用无菌取样技术。
4) If it is necessary to sample a component from the top, middle, and bottom of its container, such sample subdivisions shall not be composited for testing.
如果需要从容器的顶部、中部和底部对组件进行取样,则不得将此类分段取样混合用于检测。
5) Sample containers shall be identified so that the following information can be determined: name of the material sampled, the lot number, the container from which the sample was taken, the date on which the sample was taken, and the name of the person who collected the sample.
样品容器应予以标识,以便能够确定以下信息:被取样物料的名称、批号、样品所取自的容器、取样日期以及取样人员的姓名。
6) Containers from which samples have been taken shall be marked to show
that samples have been removed from them.
已从中取样的容器应做好标识,以表明已从这些容器中取过样。
3.1.3 21CFR§211.110 Sampling and testing of in-process materials and drugproducts.
中间物料和药品的取样与检测
(a) To assure batch uniformity and integrity of drug products, written procedures shall be established and followed that describe the in-process controls, and tests, or examinations to be conducted on appropriate samples of in-process materials of each batch. Such control procedures shall be established to monitor the output and to validate the performance of those manufacturing processes that may be responsible for causing variability in the characteristics of in-process material and the drug product. Such control procedures shall include, but are not limited to, the following, where appropriate:
为确保药品批次的均一性和完整性,应制定并遵循书面程序,描述对每批中间物料的适当样品所进行的过程控制、测试或检验。此控制程序应建立以监控生产过程的产出,并验证那些可能导致中间物料和药品特性出现变异的生产工艺的性能。此控制程序应包含但不限于以下内容(如适用):
(1) Tablet or capsule weight variation;
片剂或胶囊的重量差异;
(2) Disintegration time;
崩解时限
(3) Adequacy of mixing to assure uniformity and homogeneity;
混合的充分性,以确保均匀性和同质性;
(4) Dissolution time and rate;
溶出时间和溶出速率;
(5) Clarity, completeness, or pH of solutions.
溶液的澄清度、完整性或pH。
(6) Bioburden testing.
生物负载测试。
3.1.4 21CFR§211.122 Materials examination and usage criteria.
物料检验及使用标准。
(a) There shall be written procedures describing in sufficient detail the receipt, identification, storage,handling, sampling, examination, and/or testing of labeling and packaging materials; such written procedures shall be followed. Labeling and packaging materials shall be representatively sampled, and examined or tested upon receipt and before use in packaging or labeling of a drug product.
应制定书面程序,详细描述标签和包装材料的接收、识别、储存、处理、取样、检查及/或测试;应遵循这些书面程序。标签和包装材料应进行有代表性的取样,并在接收时以及用于药品包装或贴签之前进行检查或测试。
3.1.5 21CFR§211.134 Drug product inspection.
药品检验
(a) Packaged and labeled products shall be examined during finishing operations to provide assurance that containers and packages in the lot have the correct label.
在成品包装操作过程中,应对已包装和贴标的产品进行检查,以确保该批产品的容器和包装上贴有正确的标签。
(b) A representative sample of units shall be collected at the completion of finishing operations and shall be visually examined for correct labeling.
在成品包装操作完成时,应采集具有代表性的产品样品,并对其标签是否正确进行目视检查。
(c) Results of these examinations shall be recorded in the batch production or control records.
这些检查的结果应记录在批生产或控制记录中。
3.1.6 21CFR§211.160 General requirements.
一般要求
(a) The establishment of any specifications, standards, sampling plans, test procedures, or other laboratory control mechanisms required by this subpart, including any change in such specifications, standards, sampling plans, test procedures, or other laboratory control mechanisms, shall be drafted by the appropriate organizational unit and reviewed and approved by the quality control unit. The requirements in this subpart shall be followed and shall be documented at the time of performance. Any deviation from the written specifications,standards, sampling plans, test procedures, or other laboratory control mechanisms shall be recorded and justified.
本部分所要求的任何规格、标准、取样计划、检验程序或其他实验室控制机制的制定,包括对这些规格、标准、取样计划、检验程序或其他实验室控制机制的任何变更,均应由适当的组织单位起草,并由质量控制部门进行审核和批准。应遵循本部分的要求,并在执行时做好记录。任何偏离书面规格、标准、取样计划、检验程序或其他实验室控制机制的情况都应予以记录并说明理由。
(b) Laboratory controls shall include the establishment of scientifically sound and appropriate specifications, standards, sampling plans, and test procedures designed to assure that components, drug product containers, closures, in-process materials, labeling, and drug products conform to appropriate standards of identity, strength, quality, and purity. Laboratory controls shall include:
实验室控制应包括制定科学合理且适当的规格、标准、取样计划和检验程序,以确保组分、药品容器、密封件、中间物料、标签和药品符合有关鉴别、含量、质量和纯度的适当标准。实验室控制应包括:
(1) Determination of conformity to applicable written specifications for the acceptance of each lot within each shipment of components, drug product containers, closures, and labeling used in the manufacture, processing, packing, or holding of drug products. The specifications shall include a description of the sampling and testing procedures used. Samples shall be representative and adequately identified. Such procedures shall also require appropriate retesting of any component, drug product container, or closure that is subject to deterioration.
确定用于药品生产、加工、包装或储存的每一批组分、药品容器、密封件和标签是否符合适用的书面标准以决定其是否可接受。这些标准应包括所使用的取样和检验程序的说明。样品应具有代表性并得到充分标识。这些程序还应要求对任何可能变质的组分、药品容器或密封件进行适当的重新检验。
(2) Determination of conformance to written specifications and a description of sampling and testing procedures for in-process materials. Such samples shall be representative and properly identified.
确定中间物料是否符合书面标准,并对取样和测试程序进行说明。此类样品应具有代表性,并进行妥善标识。
(3) Determination of conformance to written descriptions of sampling procedures and appropriate specifications for drug products. Such samples shall be representative and properly identified.
确定药品是否符合取样程序的书面描述以及相应的标准要求。此类样品应具有代表性,并进行妥善标识
3.1.7 21CFR§211.166 Stability testing
稳定性测试
The written program shall be followed and shall include:
应遵循书面程序,且该程序应包括:
(1) Sample size and test intervals based on statistical criteria for each attribute examined to assure valid estimates of stability;
基于统计学标准确定的样品量和测试间隔,用于考察每一属性,以确保对稳定性进行有效评估;
(2) Storage conditions for samples retained for testing;
用于测试而留存的样品的储存条件
3.1.8 21CFR§211.170 Reserve samples.
留样
(a) An appropriately identified reserve sample that is representative of each lot in each shipment of each active ingredient shall be retained. The reserve sample consists of at least twice the quantity necessary for all tests required to determine whether the active ingredient meets its established specifications, except for sterility and pyrogen testing.
应当留存经适当标识、能代表每批活性成分发货的留样。除了无菌性和热原性测试之外,留样的数量至少应为确定该活性成分是否符合既定标准所需进行的所有测试所需数量的两倍。
3.1.9 21CFR§211.186 Master production and control records
主生产及控制记录
(9) Complete manufacturing and control instructions, sampling and testing procedures, specifications, special notations, and precautions to be followed.
完成需遵循的制造与控制说明、取样及测试程序、规格、特殊注释以及预防措施。
3.1.10 21CFR§211.194 Laboratory records
实验室记录
(a) Laboratory records shall include complete data derived from all tests necessary to assure compliance with established specifications and standards, including examinations and assays, as follows:
实验室记录应包括为确保符合既定规格和标准而进行的所有必要测试所产生的完整数据,其中包括检验和分析,具体如下:
(1) A description of the sample received for testing with identification of source (that is, location from where sample was obtained), quantity, lot number or other distinctive code, date sample was taken, and date sample was received for testing.
对接收用于测试的样品的描述,包括样品来源的标识(即取样地点)、数量、批号或其他独特的编码、取样日期以及接收样品用于测试的日期。
(2) A statement of the weight or measure of sample used for each test, where
appropriate.
适当时,说明用于每项测试的样品重量或度量情况。
3.2 EU GMPv
3.2.1 Part1Chapter4 Documentation
文件
Sampling
取样
4.25 There should be written procedures for sampling, which include the methods and equipment to be used, the amounts to be taken and any precautions to be observed to avoid contamination of the material or any deterioration in its quality.
应当有关于取样的书面程序,其中包括所使用的方法和设备、取样数量,以及为避免物料受到污染或质量下降而需遵守的任何预防措施。
3.2.2 Part1 Chapter6 Quality Control
质量控制
Quality Control is concerned with sampling, specifications and testing as well as the organisation, do cumentation and release procedures which ensure that the necessary and relevant tests are carried out, and that materials are not released for use, nor products released for sale or supply, until their quality has been judged satisfactory.
质量控制涉及取样、标准要求、测试,以及组织安排、文件记录和放行程序。这些方面确保进行必要且相关的测试,并且在物料质量被判定为合格之前,不得投入使用;在产品质量被判定为合格之前,不得进行销售或供应。
6.11 The sample taking should be done and recorded in accordance with approved written procedures that describe:
样品的采集应按照经批准的书面程序进行并记录,这些程序应描述:
i. The method of sampling
取样的方法
ii. The equipment to be used
所使用的设备
iii.The amount of the sample to be taken
所取样品的数量
iv. Instructions for any required sub-division of the sample
有关对样品进行任何必要细分的说明
v. The type and condition of the sample container to be used
待使用的样品容器的类型和状况
vi. The identification of containers sampled
已取样容器的标识
vii. Any special precautions to be observed, especially with regard to the sampling of sterile or noxious materials
需遵守的任何特殊预防措施,尤其涉及无菌或有害物料的取样方面
viii. The storage conditions
储存条件
ix. Instructions for the cleaning and storage of sampling equipment.
取样设备的清洁和储存说明。
6.12 Samples should be representative of the batch of materials or products from which they are taken. Other samples may also be taken to monitor the most stressed part of a process (e.g.beginning or end of a process). The sampling plan used should be appropriately justified and based on a risk management approach.
样品应能代表从中抽取的那批物料或产品。也可抽取其他样品来监控工艺中承受最大压力(或最关键)的部分(例如,工艺的开始阶段或结束阶段)。所采用的取样计划应充分论证,并应基于风险管理方法制定。
6.13 Sample containers should bear a label indicating the contents, with the batch number, the date of sampling and the containers from which samples have been drawn. They should be managed in a manner to minimize the risk of mix-up and to protect the samples from adverse storage conditions.
样品容器应贴有标签,标明容器内的物质、批号、取样日期以及样品的来源容器。样品容器的管理方式应能最大程度降低混淆的风险,并保护样品免受不良储存条件的影响。
3.2.3 Annex8 SAMPLING OF STARTING AND PACKAGING MATERIALS
附录 8 起始物料和包装材料的取样
Personnel who take samples should receive initial and on-going regular training in the disciplines relevant to correct sampling.
取样人员应接受与正确取样相关学科的初始培训和持续定期培训。
This training should include:
这种培训应包括:
• sampling plans
取样计划
• written sampling procedures
书面取样程序
• the techniques and equipment for sampling
取样技术和设备
• the risks of cross-contamination
交叉污染的风险
• the precautions to be taken with regard to unstable and/or sterile substances
对于不稳定和/或无菌物质需采取的预防措施
• the importance of considering the visual appearance of materials, containers and labels
考虑物料、容器和标签的外观情况的重要性。
• the importance of recording any unexpected or unusual circumstances
记录任何意外或异常情况的重要性。
Starting materials
起始物料
The identity of a complete batch of starting materials can normally only be ensured if individual samples are taken from all the containers and an identity test performed on each sample.
通常只有从所有容器中都抽取个别样品,并对每个样品都进行鉴别测试,才能确保整批起始物料的身份(即确认物料的正确属性)。
The quality of a batch of starting materials may be assessed by taking and testing a representative sample. The samples taken for identity testing could be used for this purpose.The number of samples taken for the preparation of a representative sample should be determined statistically and specified in a sampling plan. The number of individual samples which may be blended to form a composite sample should also be defined, taking into account the nature of the material, knowledge of the supplier and the homogeneity of the composite sample.
一批起始物料的质量可以通过抽取具有代表性的样品并进行测试来评估。用于鉴别测试的样品可用于此目的。为制备代表性样品而抽取的样品数量应通过统计方法确定,并在取样计划中予以明确规定。考虑到物料的性质、对供应商的了解以及混合样品的均匀性,还应确定可混合以形成混合样品的单个样品的数量。
Packaging materials
包装材料
The sampling plan for packaging materials should take account of at least the following;
包装材料的取样计划至少应考虑以下因素:
• the quantity received
接收的数量
• the quality required
所需的质量
• the nature of the material (e.g. primary packaging materials and/or printed packaging materials)
材料的性质(例如,内包装材料和/或印刷包装材料)
• the production methods, and what is known of the Quality Assurance system of the packaging materials manufacturer based on audits.
生产方法,以及根据审计所了解的包装材料制造商的质量保证体系。
The number of samples taken should be determined statistically and specified in a sampling plan.
抽取的样本数量应通过统计方法来确定,并在取样计划中明确规定。
3.2.4Annex19 Reference and Retention Samples(also Annex13 IMPs)
附录19 对照品和留样(也参照附录13 IMP)
This Annex to the Guide to Good Manufacturing Practice for Medicinal Products (“the GMP Guide”)gives guidance on the taking and holding of reference samples of starting materials, packaging materials or finished products and retention samples of finished products.
本《药品生产质量管理规范》(“《GMP 规范》”)附录就起始物料、包装材料或成品的对照样品的抽取和保存以及成品留样提供指导。
2.1 Samples are retained to fulfil two purposes; firstly to provide a sample for analytical testing and secondly to provide a specimen of the fully finished product.
留样有两个目的:第一是为分析检测提供样品,第二是留存完整成品的样本。
Samples may therefore fall into two categories
因此,样品可分为两类。
i. Reference sample: a sample of a batch of starting material, packaging material or finished product which is stored for the purpose of being analysed should the need arise during the shelf life of the batch concerned. Where stability permits, reference samples from critical intermediate stages (e.g. those requiring analytical testing and release) or intermediates, that are transported outside of the manufacturer’s control, should be kept.
参考样品:一批起始物料、包装材料或成品的样品,保存该样品是为了在有关批次的保质期内,在有需要时对其进行分析。在稳定性允许的情况下,应保留来自关键中间阶段(例如,那些需要进行分析测试和放行的阶段)的参考样品,或者是需要运输到制造商控制范围之外的中间体的参考样品。
ii. Retention sample: a sample of a fully packaged unit from a batch of finished product. It is stored for identification purposes. For example, presentation, packaging, labelling,patient information leaflet, batch number, expiry date should the need arise during the shelf life of the batch concerned.
留样:从一批成品中取出的一个完整包装单元的样品。留样是为了进行识别。例如,在有关批次的保质期内,当有需要时,留样可用于识别产品的展示形式、包装、标签、患者信息单、批号、有效期等方面。
2.3 The reference and/or retention samples serve as a record of the batch of finished product or starting material and can be assessed in the event of, for example, a dosage form quality complaint, a query relating to compliance with the marketing authorisation, a labelling/packaging query or a pharmacovigilance report
参考样品和/或留样可作为成品批次或起始物料批次的记录,并且在出现例如剂型质量投诉、与上市许可合规性相关的疑问、标签/包装方面的疑问或药物警戒报告等情况时可用于评估。
2.4 Records of traceability of samples should be maintained and be available for review by competent authorities.
应保留样品的可追溯性记录,且这些记录应可供主管部门查阅。
3.2.5 Part2;6.60 Laboratory Contro lRecords
实验室控制记录
A description of samples received for testing, including the material name or source, batch number or other distinctive code, date sample was taken, and, where appropriate, the quantity and date the sample was received for testing; -A statement of or reference to each test method used; -A statement of the weight or measure of sample used for each test as described by the method; data on or cross-reference to the preparation and testing of reference standards, reagents and standard solutions.
对收到用于检测的样品的描述,包括物料名称或来源、批号或其他独特编码、样品采集日期,以及在适当情况下,收到用于检测的样品的数量和日期;对所使用的每种检测方法的说明或引用;对按照该方法所使用的样品重量或度量的说明;关于参比标准品、试剂和标准溶液的制备和检测的数据,或对这些数据的相互参照。
3.2.6 Part2;7.33-7.35 Sampling and Testing of Incoming Production Materials
进厂生产物料的取样与检验
7.33 Samples should be representative of the batch of material from which they are taken.Sampling methods should specify the number of containers to be sampled, which part of the container to sample, and the amount of material to be taken from each container. The number of containers to sample and the sample size should be based upon a sampling plan that takes into consideration the criticality of the material, material variability, past quality history of the supplier, and the quantity needed for analysis.
样品应能代表从中抽取样品的那批物料。取样方法应规定要取样的容器数量、从容器的哪个部位取样,以及从每个容器中抽取的物料数量。要取样的容器数量和样品量应基于一个取样计划,该计划需考虑物料的关键程度、物料的变异性、供应商过去的质量记录,以及分析所需的数量。
7.34 Sampling should be conducted at defined locations and by procedures designed to prevent contamination of the material sampled and contamination of other materials.
取样应在规定的地点进行,并且要按照既定的程序操作,以防止被取样的物料受到污染,同时也要避免对其他物料造成污染。
7.35 Containers from which samples are withdrawn should be opened carefully and subsequently reclosed. They should be marked to indicate that a sample has been taken.
从其抽取样品的容器应小心开启,随后重新封闭。这些容器应做好标记,以表明已进行取样操作。
3.2.7 Part2;7.8.35 In-process Sampling and Controls
过程中取样和控制
In-process sampling should be conducted using procedures designed to prevent contamination of the sampled material and other intermediates or APIs. Procedures should be established to ensure the integrity of samples after collection.
过程中取样应采用旨在防止被取样物料以及其他中间体或原料药受到污染的程序进行。应制定程序以确保样品采集后的完整性。
3.2.8 Part2;11.52 Stability Monitoring of APIs
原料药的稳定性监测
Stability samples should be stored in containers that simulate the market container. For example, if the API is marketed in bags within fiber drums, stability samples can be packaged in bags of the same material and in smaller- scale drums of similar or identical material composition to the market drums.
稳定性样品应储存在模拟上市包装的容器中。例如,如果原料药以装在纤维桶内的袋子形式上市,稳定性样品可以包装在由相同材料制成的袋子中,并放在材料组成与上市包装的桶相似或相同的小尺寸桶内。
3.2.9 Part2;11.7 Reserve/Retention Samples
备用/留样
11.70 The packaging and holding of reserve samples is for the purpose of potential future evaluation of the quality of batches of API and not for future stability testing purposes.
备用样品的包装和保存是为了将来有可能对原料药批次的质量进行评估,而并非用于将来的稳定性测试。
3.2.10 Part2;19.8 Laboratory Controls
实验室控制
19.81 A system for retaining reserve samples of all batches should be in place. This system should ensure that a sufficient quantity of each reserve sample is retained for an appropriate length of time after approval, termination, or discontinuation of an application.
应建立一个保留所有批次备用样品的系统。该系统应确保在申请获批、终止或停止后,每个备用样品都能保留足够的数量和适当的时间。
3.3 United States Pharmacopeia
美国药典
3.3.1 General Notices
总则
Sampling is not addressed in detail in the USP as the monographs are standards not specifications and apply to any tested sample (official article).
《美国药典》中未详细阐述取样问题,因为各专论是标准而非规范,且适用于任何经测试的样品(法定物品)。
‘Standards for an article recognized in the compendia (USP–NF) are expressed in the article's monograph, applicable general chapters, and General Notices. The standards in the relevant monograph, general chapter(s), and General Notices apply at all times in the life of the article from production to expiration.’ ‘Thus, any official article is expected to meet the compendial standards if tested, and any official article actually tested as directed in the relevant monograph must meet such standards to demonstrate compliance’…in all cases, statements about whether the compendial standard is met apply only to the units tested’
在美国药典-国家处方集(USP–NF)中认可的药品标准在该药品的各专论、适用的通则章节以及凡例中阐述。从药品生产到有效期的整个生命周期内,相关专论、通则章节以及凡例中的标准始终适用。“因此,任何法定药品如果经过检测,预期应符合药典标准,并且任何按照相关专论指示实际检测的法定药品必须符合这些标准以证明其合规性”……在所有情况下,关于是否符合药典标准的表述仅适用于已检测的药品单位。
and hence
因此
‘Analytical results observed in the laboratory (or calculated from experimental measurements) are compared With stated acceptance criteria to determine whether the article conforms to compendial requirements.’
在实验室中观察到的分析结果(或从实验测量中计算得出的结果)会与规定的验收标准进行比较,以确定该药品是否符合药典要求。
However, it does discuss sampling in two above 1000 General Chapters; <1010> and <1097>.
然而,它确实在两篇编号大于 1000 的通则章节(即<1010>和<1097>)中讨论了取样问题。
3.3.2 General Chapter<1010>ANALYTICAL DATA—INTERPRETA TIONAND
TREATMENTviii
通则章节<1010>分析数据——解释与处理
Effective sampling and randomization are important considerations in mitigating the impact of bias.Sampling is performed after the study has been designed and constitutes the selection of test articles within the structure of the design. How to attain such a sample depends entirely on the question that is to be answered by the data. When possible, use of a random process is considered the most appropriate way of selecting samples.
有效的取样和随机化是减轻偏差影响的重要考量因素。取样工作在研究设计完成之后进行,且取样是在研究设计架构内对测试物品的选取。如何获得这样的样本完全取决于需要通过数据来解答的问题。在可能的情况下,采用随机程序被认为是选择样本最合适的方式。
The most straightforward type of random sampling is called simple random sampling. However,sometimes this method of selecting a random sample is not desirable because it cannot guarantee equal representation across study factors. The design of a study to release manufactured lots might incorporate factors such as selected times, locations, or parallel manufacturing streams (e.g.,multiple filling lines). In this case a stratified sample whereby units are randomly selected from within each factor can be utilized.
最直接的随机取样类型被称为简单随机取样。然而,有时这种选择随机样本的方法并不理想,因为它无法保证在研究因素中具有同等的代表性。为放行生产批次而进行的研究设计可能会纳入诸如选定的时间、地点或并行的生产流程(例如,多条灌装生产线)等因素。在这种情况下,可以采用分层抽样,即从每个因素中随机选择样本单元。
Regardless of the reason for taking a sample, a sampling plan should be established to provide details on how the sample is to be obtained to ensure that it is representative of the entirety of the population of interest.
无论出于何种原因进行取样,都应制定取样计划,详细说明如何获取样本,以确保其能代表所关注总体的全部情况。
3.3.3 GeneralChapter<1097>BULK POWDE RSAMPLING PROCEDURES
通则章节<1097>散装粉末取样程序
The goals of this chapter are to provide guidance on bulk powder sampling procedures, identify important bulk powder sampling concepts, and collect a knowledge base of useful practices and considerations that can lead to the ideal physical sampling of bulk powder materials.
本章的目的是为散装粉末取样程序提供指导,明确重要的散装粉末取样概念,并收集有用的实践和注意事项知识库,以实现散装粉末材料的理想物理抽样。
The primary difficulty in acquiring a representative sample is that the size of the sample for measurement, typically a few milligrams to grams, must be withdrawn from a large population on the order of hundreds to thousands of kilograms. The few milligrams analysed in a laboratory must be taken from a large population of particles in a warehouse in such a manner that the measurement sample is representative of all the particles in the lot.
获取具有代表性的样品的主要困难在于,用于测量的样品规模通常为几毫克到几克,而这些样品必须从数量达数百到数千千克的大量物料中提取。在实验室中分析的几毫克样品,必须以某种方式从仓库中的大量颗粒物料中获取,从而使测量样品能够代表该批次的所有颗粒。
A typical sampling strategy consists of two basic steps: (1) the primary or gross sample, followed by(2) the secondary sample, which reduces the primary sample to a size that is suitable for laboratory measurement. In short, the goal is to select from the lot a quantity of material suitable for measurement without significantly changing the attribute for which one is sampling.
一个典型的取样策略包含两个基本步骤:(1)获取初始或粗样,接着(2)获取二次样,即将初始样品缩减至适合在实验室测量的规模。简而言之,目标是从批次中选取适合测量的物料数量,同时不会显著改变进行取样所针对的属性。
In parallel with the sample size reduction, sample size calculations must be done in such a way that the sampling strategy has sufficient statistical power to determine whether the attributes of interest lie within the specification ranges with a reasonable degree of certainty. Each step must be done correctly, or the sampling strategy as a whole will not provide a sample that is representative of the
original population.
在减小样品规模的同时,必须进行样品量的计算,以使取样策略具备足够的统计效力,从而能够在合理的确定程度下判定所关注的属性是否处于规定的范围之内。每一个步骤都必须正确执行,否则作为一个整体的取样策略将无法提供能够代表原始总体的样品。
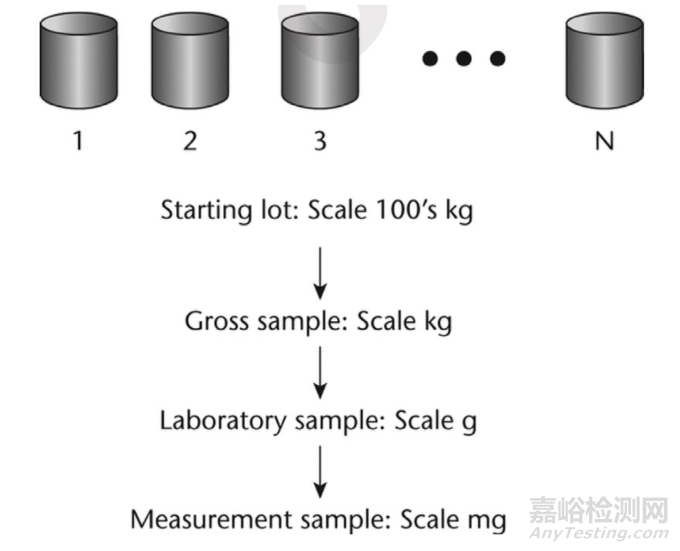
Figure 3 Sampling strategy that will help to minimize sampling errors
图 3 有助于将取样误差降至最低的取样策略
3.4 WHO Guidance for sampling of pharmaceutical products and related materials
WHO 药品及相关物料的取样指导意见
1. Introduction
介绍
1.1 General considerations
一般考虑因素
1.2 Glossary
术语表
1.3 Purpose of sampling
取样的目的
1.4 Classes and types of pharmaceutical products and related materials
药品及相关物料的类别和类型
1.5 Sampling facilities
取样设备
1.6 Responsibilities for sampling
取样的职责
1.7 Health and safety
健康与安全
2. Sampling process
取样过程
2.1 Preparation for sampling
取样准备
2.2 Sampling operation and precautions
取样操作及注意事项
2.3 Storage and retention
储存和保留
3. Regulatory issues
法规问题
3.1 Pharmaceutical inspections
药品检查
3.2 Surveillance programmes
监督项目
4. Sampling on receipt (for acceptance)
收货时取样(用于验收)
4.1 Starting materials
起始物料
4.2 Intermediates in the manufacturing process and bulk pharmaceutical products
生产过程中的中间体以及原料药
4.3 Finished products
成品
4.4 Packaging materials (primary and secondary)
(一级和二级)包装材料
5. Sampling plans for starting materials, packaging materials and finished products
起始物料、包装材料和成品的取样计划
5.1 Starting materials
起始物料
5.2 Packaging materials
包装材料
5.3 Finished products
成品
Bibliography
参考资料
Appendix 1 Types of sampling tools
取样工具的类型
Appendix 2 Sample collection form
样品采集表
Appendix 3 Steps to be considered for inclusion in a standard operating procedure
附录 3 纳入标准操作规程时需考虑的步骤
Appendix 4 Examples of types of containers used to store samples of starting materials and bulk products
附录 4 用于储存起始物料和大宗产品样品的容器类型示例
Appendix 5 Examples of use of sampling plans n, p and r. See section 6.3 for more details.
3.5 FDA Guidance Investigating Out-of-Specification (OOS) Test Results for
Pharmaceutical Production, May 2022
《2022 年 5 月美国食品药品监督管理局(FDA)药品生产中超出规格(OOS)测试结果调查指南》
Retesting and resampling are a critical aspects of this guidance.
重新测试和重新采样是该指南的关键方面
Part of the investigation may involve retesting of a portion of the original sample. The sample used for the retesting should be taken from the same homogeneous material that was originally collected from the lot,tested, and yielded the OOS results. For a liquid, it may be from the original unit liquid product or composite of the liquid product; for a solid, it may be an additional weighing from the same sample composite prepared for the original test.
调查的一部分可能涉及对原始样品的一部分进行重新测试。用于重新测试的样品应取自最初从批次中采集、经过测试并得出不合格(OOS)结果的同一均匀物料。对于液体,它可能取自原始的单位液体产品或液体产品的混合样;对于固体,它可能是从为原始测试准备的同一样品组合中额外称取的部分。
3.6 GOOD Samples: Guidance on Obtaining Defensible Samples
良好样本:获取可靠样本的指南
The FDA regulates food as well as drugs, In October 2015, the Sampling and Sample Handling Working Group of the Association of American Feed Control Officials (AAFCO) developed a guidance by a consortium, led by FDA, entitled GOODSamples: Guidance on Obtaining Defensible Samples.This 83 page guidance had the objective to “improve analytical data equivalency among state,federal and local agencies ... Because analytical data is only as good as the quality of thesample”.
美国食品药品监督管理局(FDA)既监管食品也监管药品。2015 年 10 月,美国饲料控制官员协会(AAFCO)的取样和样品处理工作组在 FDA 的领导下,由一个联合体制定了一份名为《优质样本:获取可靠样本的指南》的文件。这份长达 83 页的指南旨在“提高州、联邦和地方机构之间分析数据的等效性……因为分析数据的质量仅取决于样本的质量” 。
A subsequent document in 2018, GOOD Test Portions: Guidance On Obtaining Defensible Test Portions (70 pages) was developed and contains detailed information on the TOS in respect of the error structure and sampling methodology for foodstuffs.
随后在 2018 年的一份文件《优质测试部分:获取可靠测试部分的指南》(70 页)得以制定,其中包含了关于食品的测试样本(TOS)在误差结构和取样方法方面的详细信息。
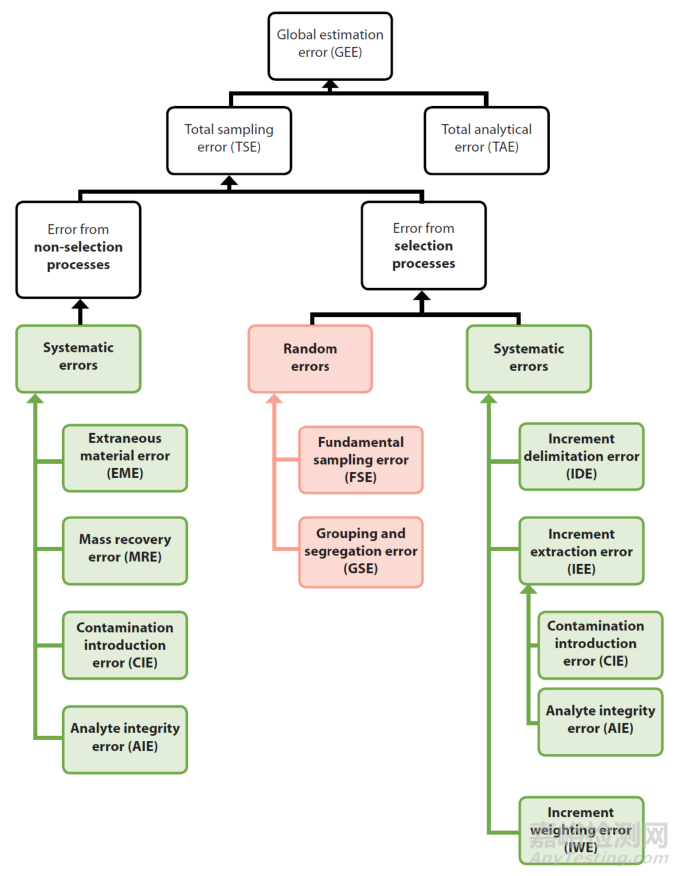
Figure 4 Categorization of errors contributing to total sampling error and relationship to global estimation error [from figure 4 of reference
图 4 对导致总取样误差的误差进行分类以及与总体估计误差的关系
4 Theory of Sampling
抽样理论
The theory of sampling (TOS) has been developed, primarily by Dr Pierre Gy, between 1950 and the early 2000s. In Gy theory, correct sampling is defined as a sampling scenario in which all particles have the same probability of being included in the sample. The theory was primarily developed for the mining industries and large scale sampling problems of large quantities of bulk materials as to how to generate a representative sample.
抽样理论(TOS)主要是由皮埃尔·吉博士在 1950 年至 21 世纪初期间提出的。在吉的理论中,正确的抽样被定义为这样一种抽样情形:所有颗粒都有相同的被纳入样本的概率。该理论主要是为采矿业以及大量散装物料的大规模抽样问题而发展起来的,旨在解决如何获取有代表性的样本这一问题。
These two books are high level monographs and statistically very detailed. More recently, an introductory book has been published by Esbensen.
这两本书是高水平的专著,并且在统计学方面非常详尽。最近,埃斯本森出版了一本入门书籍。
A more accessible and less statistical book on the sampling and weighing of bulk solids was published in 1985xvii. In addition, two older books may also be found useful
1985 年出版了一本关于散装固体抽样和称重的书籍,该书更容易理解,且统计学内容较少。此外,还有两本较早的书籍可能也有用处。
The published literature is sparse with respect to pharmaceuticals and related materials. Representative sampling has been briefly addressedxx to understanding the differences between convenience, target, and self-selected samples. In addition, compositing of samples has also been discussed by Torbeckxxi because compositing samples is appropriate under certain circumstances but raises caveats on how and when it should be applied.
就药品及相关材料而言,已发表的文献数量稀少。对于代表性抽样,只是简略提及了它对于理解便利抽样、目标抽样和自选择抽样之间差异的作用。此外,托尔贝克也讨论过样本混合的问题,因为在某些情况下样本混合是合适的,但在如何应用以及何时应用方面需要注意一些事项。
The best practical application of sampling practices is the WHO guidances for sampling of pharmaceutical products and related materials ix. However, more details regarding the sampling plan sections are given in Chapter 6.
抽样实践的最佳实际应用是世界卫生组织关于药品及相关材料抽样的指导原则。不过,有关抽样计划部分的更多详细内容在第6 章中给出。
5 Nomenclature of sampling
抽样的术语
Consistent sampling and testing nomenclature are critical for data integrity and compliance with specifications and regulatory standards. Currently, there is a lack of agreement between the major sources of guidance both within the pharmaceutical industry and between other regulated sectors The International Union of Pure and Applied Chemistry (IUPAC) made nomenclature recommendations for sampling in analytical chemistry in 1990xxii. This paper was referenced in the 2023 IUPAC Orange book.
统一一致的抽样和测试术语对于数据完整性以及符合规范和监管标准至关重要。目前,制药行业内部以及其他受监管部门之间的主要指导来源在这方面缺乏共识。国际纯粹与应用化学联合会(IUPAC)在 1990 年就分析化学中的抽样问题提出了有关术语的建议。2023 年的 IUPAC “橙皮书” 中引用了这篇论文。
ASTM Standard Guide for Defining the Test Result of a Test Method, E2282-14(2019) has also produced a nomenclature guide.
美国材料与试验协会(ASTM)的《定义测试方法的测试结果的标准指南》(E2282-14(2019))也制定了一份术语指南。
The similarities and differences between literature references have been discussed.
当讨论文献参考之间的异同点时,以下几个方面可能会被涉及到:
The four key documents for a particular material or product are;
文献参考资料之间的异同点已被讨论过了。
1. Statistical Sampling Plan
统计取样计划
2. Sampling Procedure
去样程序
3. Sampling Protocol
取样规程
4. Sampling Record
取样记录
The relationship between these documents are shown in Figure 1 and some proposed terms and definitions listed in Table 1.
这些文件之间的关系如图 1 所示,一些提议的术语和定义列于表 1 中。
Table 1 Proposed terms and definitions
提议的术语和定义
|
|
|
|---|---|
|
统计取样计划 |
A predetermined procedure for the selection, withdrawal, preservation and preparation of the portions to be removed from a population as samples. - 样本是从大量材料中选取的任何一部分材料。样本类型由特定的限定词来界定,例如随机样本,每种样本类型都有其自身的定义。 A sample is any portion of material selected from a larger quantity of material. The sample type is identified by specific qualifiers, e.g. random sample, with their own definitions. - 其目的是在实际约束条件下,将根据样本估算出的属性与批次的实际属性之间的差异降至最低,并在统计学基础上限制或控制抽样操作所产生的不确定性。 The intent is to minimize the difference between the properties as estimated from a sample and the actual properties of the lot, and to limit or control the uncertainty generated by the sampling operation on a statistical basis, all within practical constraints. - 在实际限制条件下,基于统计学原理进行抽样操作。 sampling operation on a statistical basis, all within practical constraints. - 终点是一个用于测试的具有代表性的样本,该样本应能充分反映总体所关注的属性(特征)。 The end point is a representative sample for testing that can be expected to adequately reflect the properties of interest (Characteristics) of the parent population. |
|
特征 |
A property or attribute of a material that is to be measured or observed. The ISO term is measurand. |
|
抽样不确定度(均匀性) |
That part of the error associated with using only a fraction of the material population to be sampled and extrapolating to the whole as distinct from analytical or testing error. It arises from a lack of homogeneity in the material population. - 抽样误差可以通过对样本增量进行重复操作以及多次测定来确定,并且(或者)在适当的情况下通过减小颗粒大小来确定。 Sampling error may be determined by replication of sample increments and their multiple determinations or/and by reducing the particle size if appropriate. - 均匀性是指一种属性、特征或特性在一定数量的材料中均匀分布的程度。非均匀性(均匀性的反面)是抽样误差的决定因素。 Homogeneity is the degree to which a property, characteristic or attribute is uniformly distributed throughout a quantity of material. The degree of heterogeneity (the opposite of homogeneity) is the determining factor of sampling error. |
|
抽样过程(定义) |
Static sampling: The composition of the parent material can be considered as permanent with respect to position in space and stable in time such that the only variable is the sampling position within the space occupied by the consignment, lot or batch. - 动态抽样:如果母体材料几乎总是随时间变化(具有动态性),并且在任何时刻抽取母体材料的一部分都仅反映了该时刻以及特定位置的状态,例如从混合器中获取的过程控制样本。 Dynamic sampling: If the parent material is almost always changing with respect to time (dynamic) and the removal of a portion of the parent material at any instant reflects only a state at that time and at a particular site e.g. In process control samples from a mixer. |
|
|
Sample increment:Each of the discrete, identifiable portions of material removed from a population which can be individually tested. |
|
|
Random sample:The sample so selected that any portion of the population has an equal (or known) chance of being chosen. |
|
|
Representative sample:A sample resulting from a sampling plan. |
|
|
Reference sample:A representative sample of a batch of starting material, packaging material or finished product which is stored for the purpose of being analysed should the need arise during the shelf life of the batch concerned. |
|
|
Retention sample:A sample of a fully packaged unit from a batch of finished product. It is stored for identification purposes. For example, presentation, packaging, labelling, patient information leaflet, batch number, expiry date should the need arise during the shelf life of the batch concerned. |
|
|
Stratified sample:A sample consisting of portions obtained from identified subparts (strata) of the parent population. Within each stratum, the samples are to be taken randomly. |
|
|
Sampling Procedure:The complete sampling operations to be performed on a defined material for a specific purpose. |
|
|
Sampling Protocol:A detailed written description of the sampling procedure to be carried out for a specific consignment, lot or batch. |
|
取样记录 |
Documented evidence of the sampling operations carried out on a particular material for a defined purpose. The sampling record should contain the batch number, date and place of sampling, reference to the sampling protocol used, a description of the containers and of the materials sampled, notes on possible abnormalities, together with any other relevant observations, and the name(s) and signature(s) of the sampler(s) in accordance with ALCOA++ principles. [Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, Available, Traceable] |
6 The Sampling Process
抽样过程
The sampling process is comprised of two distinct phases: design and implementation.
抽样过程由两个截然不同的阶段组成:设计阶段和实施阶段。
1. In the design phase, the application of statistical principles is expected to supply a statistical sampling plan to determine the acceptability of a lot, the magnitude of a property, or the quantity of a constituent, within a specified degree of variability with a stated degree of confidence, the document describes,
在设计阶段,统计原理的应用旨在提供一个统计抽样计划,以便在规定的变异性程度和既定的置信度下,确定一批产品的可接受性、一种特性的量值或者一种成分的数量。该文件对此进行了描述。
2. The implementation phase of sampling involves the physical realization of the statistically designated portions and the removal and preparation of them—an operation easier to state than to perform.
抽样的实施阶段涉及对按统计指定部分的实际获取,以及对这些部分的提取和准备工作, 这一操作说起来容易,做起来难。
6.1 Statistical Sampling Plans
统计抽样计划
A statistical sampling plan is a high-level master document containing all necessary definitions and structures to ensure that a representative sample from a given material population is available for testing in the laboratory. This is a critical document because sampling is always an error-generating process.
统计抽样计划是一份高层次的主要文件,它包含了所有必要的定义和架构,以确保能从给定的物料总体中获取具有代表性的样本,用于实验室检测。这是一份至关重要的文件,因为抽样始终是一个会产生误差的过程。
The suggested main terms and definitions are shown in Table 1.
建议的主要术语和定义见表1。
The concept of sampling populations is straight forward, in theory, in that a representative sample is taken from the population and examined for compliance with acceptance criteria be they attributes(e.g. presence of defects) or variables (e.g. dimension within tolerance). On the basis of the finding the level of non-conformances in the representative sample, take a calculated risk of accepting or rejecting the batch (Population) using Acceptance Sampling.
抽样总体的概念从理论上来说是直接明了的,即从总体中抽取一个有代表性的样本,并检查该样本是否符合验收标准,这些标准可以是属性方面的(例如是否存在缺陷),也可以是变量方面的(例如尺寸是否在公差范围内)。基于对有代表性样本中不符合项水平的调查结果,运用验收抽样方法来承担接受或拒收该批次(总体)的计算风险。
There will always be a chance that;
总会存在这样一种可能性:
• good material will be rejected; called the Producer’s risk
合格的材料会被拒收,这被称为生产方风险。
• bad material will be accepted; called the Consumer’s risk
不合格的材料会被接受,这被称为使用方(消费者)风险。
6.2 Acceptance Sampling Plans
验收抽样计划
Acceptance sampling plans are available for attributes and variables. The most commonly used in the pharmaceutical industry is for attributes.
验收抽样计划可用于属性抽样和变量抽样。在制药行业中,最常用的是属性抽样计划。
The mathematical model for sampling without replacement is the hypergeometric distribution whose cumulative distribution function (CDF) is given by
无放回抽样的数学模型是超几何分布,其累积分布函数(CDF)由以下给出 :
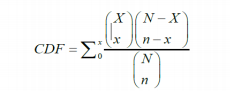
Where xis the number of occurrences of non-conformities found in the sample and nis the number of samples taken from the population. Nis the batch size and Xis the number of non-conformances in it. The () terms are the binomial coefficients.
其中x是在样本中发现的不合格品的数量,n是从总体中抽取的样本数量。N是批次大小,X是其中不合格品的数量。( )项是二项式系数。
What is required is an Operational Characteristic (OC) curve based upon a sample size, n,a defined acceptance criterion, (How many defects to accept), and the batch
size.
所需的是一条基于样本量n、规定的验收标准(可接受的缺陷数量)以及批次规模的操作特性(OC)曲线。
This OC curve is a plot of the probability of accepting a batch containing a specified % non-conformance (Y axis), based upon the hypergeometric distribution, and the % non-conformances(X axis).
这条操作特性(OC)曲线是基于超几何分布,以接受含有特定不合格品百分比的批次的概率(纵轴)与不合格品百分比(横轴)所绘制的图表。
Fortunately, predetermined ISO standard sampling plans are available for use in laboratories for attributesxxviii and variablesxxix which do not require the calculation of the CDF!
幸运的是,对于属性抽样和变量抽样,实验室有预先确定的国际标准化组织(ISO)标准抽样计划可供使用,这些计划无需计算累积分布函数(CDF)!
By way of a simple example to illustrate an OC curve, suppose, we have a batch size Nof 1000 items,and decide to take a sample size (n) of 50 intending that we will accept the batch if we have one or less defects after examining n samples otherwise we will reject it.
通过一个简单的例子来阐释操作特性(OC)曲线。假设一批产品的批量N为1000件,我们决定抽取样本量=50。如果在检验了n个样本后,发现的缺陷数量为1个或更少,我们就接受这批产品,否则就拒收该批产品。
We can therefore calculate the OC curve using the CDF function, using Excel, and interpret it.
因此,我们可以使用Excel中的累积分布函数(CDF)来计算操作特性(OC)曲线,并对其进行解释。
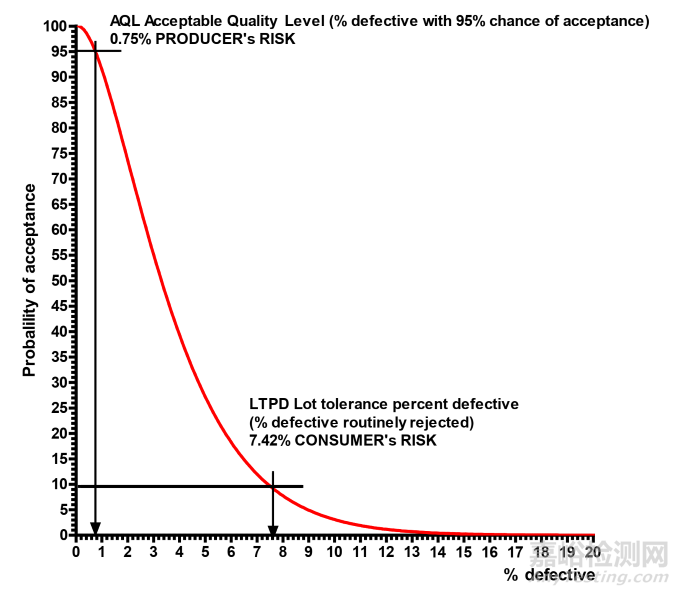
Figure 5 Example Operating Characteristic Curve
图5 操作特性曲线示例
The AQL with a 95% chance of acceptance gives a producer’s risk of rejection as 0.75%. However,the consumer’s risk is 10 times higher! The LTPD is the percentage defective with a 10% chance of acceptance. This is the probability of falsely accepting a bad lot!
可接受质量水平(AQL)下,接受的可能性为 95%时,生产方被拒收的风险为 0.75%。然而,使用方的风险要高出 10 倍!极限质量水平(LTPD)是指当接受概率为 10%时的不合格品百分比。这就是误收不良批次的概率!
This guidance provides only a simplified overview of acceptance sampling. There is an excellent book which covers acceptance sampling in great detail.
这份指南仅仅提供了验收抽样的一个简化概述。有一本非常优秀的书籍对验收抽样进行了极为详细的阐述。
In practice, we select a sampling plan based upon a batch size and an acceptance quality limit (AQL)based upon the risk associated with a defect or non-conformance using the sampling tables from ISO 2859-1 for attributes.
在实际操作中,我们根据批次大小以及基于与缺陷或不符合项相关的风险所确定的可接受质量限(AQL),并使用 ISO 2859-1 中针对属性抽样的抽样表来选择抽样计划。
The risk-based strategy depends upon the risk level; Critical, Major or Minor (e.g. cosmetic defects). Let us assume one example for each risk level. The corresponding AQLs for the risk levels are 0.010%, 0.065% and 4.0% respectively. Let us also assume that the corresponding batch sizes are 300,000, 100,000 and 1000.
基于风险的策略取决于风险等级,即关键等级、主要等级或次要等级(例如外观缺陷)。让我们为每个风险等级举一个例子。对应这些风险等级的可接受质量限(AQL)分别为 0.010%、0.065%和 4.0%。我们同时假设相应的批次大小分别为 300,000、100,000 和 1000 。
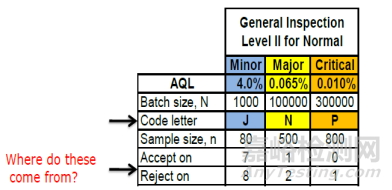
Figure 6 Example AQLs Sample Sizes & Acceptance Criteria
示例可接受质量限、样本量和验收准则
The sampling tables do not use exact sample sizes but sample size ranges as an approximation for any given inspection level. In this example, Level II (normal) has been selected which would be the usual practice.
抽样表并不采用精确的样本量,而是使用样本量范围来近似表示任何给定的检验水平。在这个例子中,选择了检验水平 II(正常检验水平),这也是通常的做法。
The coloured sample size code letter for the batch size is shown in Figure 6.
针对该批次规模的带颜色的样本量编码如图 6 所示。
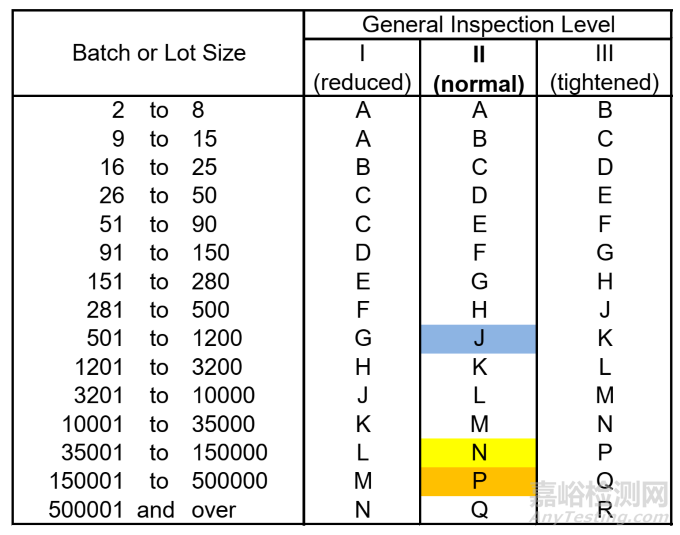
Figure 7 Allocation of sample size range code letter to Level II (normal)
图7 检验水平II(正常检验水平)的样本量范围编码分配情况
The accept (Ac)/reject (Re) acceptance numbers depend on the sample size range code letter and the selected AQL. The representative sample sizes to be examined are linked to the sample size range code letter also.
接收(Ac)/拒收(Re)的接收数取决于样本量范围编码字母以及所选的可接受质量限(AQL)。要检验的代表性样本量也与样本量范围编码字母相关联。
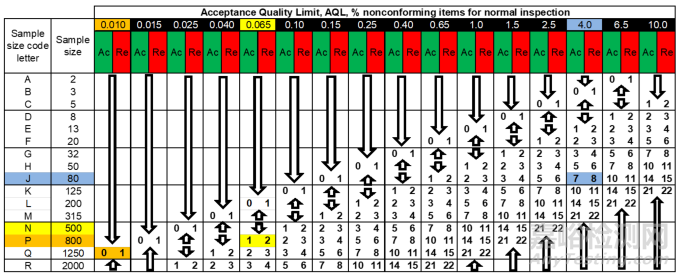
Figure 8 The accept (Ac)/reject (Re) acceptance numbers for inspection Level II (normal) as a function of AQL.
检验水平 II(正常检验)下,作为可接受质量限(AQL)函数的接收(Ac)/拒收(Re)接收数
However, there is flexibility to switch between reduced and tightened inspection Levels (I & III)depending on the ongoing supplier performance. After 30 conforming lots or batches, a reduction of testing is warranted and reduced inspection level I is able to be implemented. However, if a lot fails then Level II is reinstated. If normal inspection (Level II) operation find that 2 of 5 inspections fail the Level III tightened inspection is implemented. If a total of 5 lots are rejected then inspection should be discontinued until the supplier rectifies the quality issue.
然而,根据供应商的持续表现,可以灵活地在放宽检验和加严检验(检验水平I和检验水平III)之间切换。当有30批合格产品时,就有理由减少检验量,并可实施放宽检验水平I。不过,如果有一批产品不合格,就要恢复到检验水平II。如果在正常检验(检验水平II)操作中发现5次检验中有2次不合格,就要实施加严检验水平III。如果总共有5批产品被拒收,那么就应该停止检验,直到供应商纠正质量问题为止。
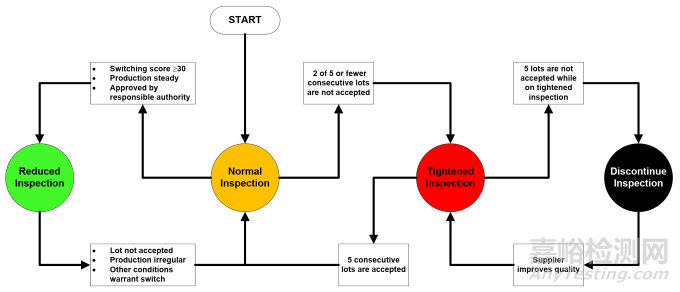
Figure 9 Quality performance; Switching rules
图 9 质量表现;转换规则
6.3 WHO guidelines; three reduced sampling plans for starting materials
WHO 针对起始物料的三种简化抽样方案
Pages 61 to 93 of this guideline discusses three reduced sampling plans for starting materials; the n,pand rplans. The use of any reduced sampling plan needs to be justified based upon data.
本指南的第61页至第93页讨论了针对起始原料的三种简化抽样方案,即n方案、p方案和r方案。任何简化抽样方案的使用都需要有数据作为依据来证明其合理性。
6.3.1 The n plan
The most commonly used plan is the n plan, more commonly called the ‘Root N +1 reduced sampling plan. The statistical aspects have been discussed by Torbeck in 2009. The n plan ‘assumes a uniform material from a recognized source where there is a high degree of confidence in the source ’. However, WHO makes a strong warning about the use of this plan.
最常用的方案是 n 方案,它更普遍地被称为 “根号 N 加 1 简化抽样方案”。托尔贝克在 2009 年探讨了该方案的统计学方面的内容。n 方案 “假定所采用的材料来自可认可的来源,且对该来源有高度的信任,并且物料性质均一”。然而,WHO强烈警示了使用该方案的情况。
The n plan is not recommended for use by control laboratories of manufacturers who are required to analyse and release or reject each received consignment of the starting materials used to produce a drug product.
对于那些需要对所接收的用于生产药品的起始物料进行分析并决定放行或拒收的生产企业的质量控制实验室而言,不建议使用n方案。
In 5.1.1, it continues;
在 5.1.1 部分,内容继续写道:
The “n plan” should be used with great caution and only when the material to be sampled is considered uniform and is supplied from a recognized source. Samples can be withdrawn from any part of the container (usually from the top layer).
“n方案”应极其谨慎地使用,并且只有在待抽样的物料被认为是性质均一的,且由一个已认可的来源提供的情况下才可使用。样品可以从容器的任何部位抽取(通常从顶层抽取)。
N is the number of sampling units in the consignment. The value of n is obtained by simple rounding. A minimum number of containers needs to be sampled, e.g. if N is less than or equal to 4, then every container is sampled.
N是该批货物中的抽样单元数量。n的值通过简单的取整得到。需要对一定数量的容器进行抽样,例如,如果 N 小于或等于 4,则需对每个容器进行抽样。
6.3.2 The p plan
p 方案
In 5.1.2 it describes the p plan noting the caveat
在 5.1.2 中描述了该计划,并提到了相关的注意事项。
The “p plan” may be used when the material is uniform, is received from a recognized source and the main purpose is to test for identity. The p plan is based on the formula p = 0.4 *Root N, where N is the number of sampling units. The figures for p are obtained by rounding up to the next highest integer.
当物料性质均一,且来自已认可的来源,同时主要目的是进行鉴别检验时,可以使用“p方案”。“p方案”基于公式p=0.4×根号N,其中N是抽样单元的数量。p的数值通过向上取整到下一个最高整数得到。
According to this plan, samples are taken from each of the N sampling units of the consignment and placed in separate sample containers.
根据这个方案,从该批货物的 N 个抽样单元中的每一个抽取样本,并将其放置在单独的样品容器中。
These original samples are transferred to the control laboratory, visually inspected and tested for identity (a simplified method may be used). If the results are concordant, p final samples are formed by appropriate pooling of the original samples.
这些原始样品被送往质量控制实验室,进行目视检查并开展鉴别检验(可以使用简化方法)。如果检验结果一致,通过对原始样品进行适当的合并来形成 p 个最终样品。
6.3.3 The r plan
r 方案
In 5.1.3, it describes the r plan
在 5.1.3 部分,它描述了 r 方案。
The “r plan” may be used when the material is suspected to be nonuniform and/or is received from a source that is not well known. The r plan may also be used for herbal medicinal products used as starting materials. This plan is based on the formula r = 1.5*Root N, where N is the number of sampling units. The figures for r are obtained by rounding up to the next highest integer.
当物料被怀疑性质不均一,以及/或者物料来自一个不太知名的来源时,可以使用 “r 方案”。“r方案” 也可用于作为起始原料的草药制品。该方案基于公式 r = 1.5×根号 N ,其中 N 是抽样单元的数量。r 的数值通过向上取整到下一个最高整数得到。
Samples are taken from each of the N sampling units of the consignment and placed in separate sample containers. These original samples are transferred to the control laboratory and tested for identity. If the results are concordant, r samples are randomly selected and individually subjected to testing. If these results are concordant, the r samples are combined for the retention sample.
从该批货物的N个抽样单元中的每一个抽取样品,并将其放置在单独的样品容器中。这些原始样品被送往质量控制实验室并进行鉴别检验。如果检验结果一致,随机选取r个样品,并分别对它们进行检测。如果这些检测结果一致,则将这 r 个样品合并作为留样。
Illustrative examples of these 3 plans are given in Figure 10.
这三种方案的示例在图10中给出。
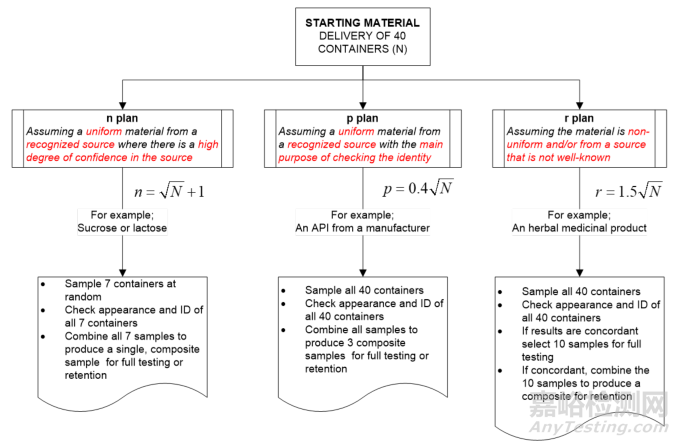
Figure 10 Examples of the WHO n, p & r plans for a consignment of 40 container
对于一批包含 40 个容器的货物,WHO 的 n 方案、p 方案和 r 方案的示例
7 Sampling Procedure & Protocol
取样程序&方案
The parent material (population) to be sampled is a consignment, batch or lot, uniquely identified,and is specified in the procedure and uniquely recorded as part of the procedure and the protocol.
待取样的母体物料(总体)是一批货物、一个批次或一批产品,需进行唯一标识,并在抽样程序中加以明确规定,且作为取样程序和方案的一部分进行唯一记录。
The collection of one or more increments or units taken from a population as required by the material specific sampling procedure and the batch/material specific sampling protocol and suitably identified.
根据特定物料的取样程序以及特定批次/物料的取样方案的要求,从总体中采集一个或多个份样或抽样单元,并进行适当标识。
The overall sampling process is driven by the sampling protocol and the requirements of the statistical sampling plan (SSP) for a particular material. This protocol contains the complete sampling operations to be performed on a defined material for a specific purpose and is traceable to the SSP.
整个取样过程由取样方案以及针对特定物料的统计抽样计划(SSP)的要求所驱动。该方案包含了为特定目的而对特定材料需执行的完整取样操作,并且可追溯至统计抽样计划(SSP)。
These increments may be combined (composited) to form a combined sample if specified in the protocol. A sufficient and representative amount sample for testing, of at least twice the amount required for testing, that can be expected to adequately reflect the characteristics of the parent population should be taken.
如果取样方案有规定,这些份样可以合并(组成混合样)。应抽取足够且具有代表性的样本用于检测,其数量至少为检测所需量的两倍,且该样本应能充分反映母体总体的特征。
7.1 In-process sampling
过程中取样
Much of the sampling effort in companies is focused on materials received and finished products. The importance of a constant structured approach to in-process sampling is often overlooked.
公司的取样工作大部分集中在进货物料和成品上。然而,对于过程取样采用持续的、结构化方法的重要性常常被忽视。
Recently FDA have found it necessary to issue a specific draft guidance in this area.
最近,FDA 认为有必要在这一领域发布一份特定的指导草案。
In section III. GENERAL CONSIDERATIONS FOR IN-PROCESS SAMPLING AND TESTING, it states;
在第三节“过程取样和测试的一般注意事项”中,其陈述如下:
‘The determination of whether in-process controls, and tests, or examinations meet the regulatory requirements in §211.110 primarily depends on the nature of the drug product (e.g., dosage form) and the type of process used by the manufacturer .’
“对于过程控制、测试或检查是否符合第 211.110 条的监管要求的判定,主要取决于药品的性质(例如剂型)以及制造商所采用的生产过程类型。”
And
并且
‘In addition to identifying which critical quality attributes and in-process material attributes to monitor, the manufacturer should define and justify where and when the proposed in-process controls, and testing, or examinations that are used to monitor those attributes should occur.’
“除了确定需要监测的关键质量属性和过程物料属性之外,制造商还应明确说明并提供合理依据,确定在哪些位置以及何时进行用于监测这些属性的过程控制、测试或检查。”
And specifically
具体而言
‘The manufacturer should employ a scientifically sound and appropriate sampling and testing strategy for quality attributes at appropriate points in the process that are adequate to ensure drug product quality. The manufacturer should employ time-based sampling plans for quality attributes, where appropriate.’
制造商应在生产过程的适当环节,针对质量属性采用科学合理且恰当的取样和测试策略,以充分确保药品质量。在适当情况下,制造商应针对质量属性采用基于时间的取样计划。
‘Although in-process controls, and tests, or examinations of in-process materials are required, sampling does not necessarily require steps for physically removing in-process materials to test their characteristics.Innovative technologies allow in-line, at-line, or on-line measurements in place of physical sample removal for laboratory testing.’
“尽管需要对过程物料进行过程控制、测试或检查,但取样并不一定需要通过实际取出过程物料来测试其特性。创新技术允许进行在线、线旁或在线测量,以替代取出实物样品进行实验室测试。”
‘In batch manufacturing of a solid oral drug product, blend uniformity should typically be assessed before in-process materials continue to the compression step. However, in continuous manufacturing, a manufacturer could conduct sampling and testing at an appropriate point in the process (e.g., at the tablet press feed frame or after compression) to evaluate the adequacy of mixing to ensure batch uniformity and homogeneity.’
“在固体口服药品的批量生产中,通常应在过程物料进入压片步骤之前评估混合均匀性。然而,在连续生产中,制造商可以在生产过程的适当阶段(例如,在压片机进料框架处或压片之后)进行取样和测试,以评估混合的充分性,从而确保批次的均匀性和均一性。”
8 Sampling Record
取样记录
The sampling record is the documented evidence of the actual sampling operations carried out on a particular material for a defined purpose.
取样记录是为特定目的对特定物料实施的实际取样操作的书面证明文件。
The sampling record should contain;
取样记录应包含:
• a unique identifier for the parent material
母体物料的唯一识别码(或唯一标识)
• date, time and place of sampling
取样日期、时间和地点
• reference to the sampling protocol used and, by inference, the SST
提及所使用的取样方案,并且由此推断出统计抽样计划(SST)。
• a description of the containers, with labelling instructions, of the materials sampled
对所抽取材料的容器的描述,包括贴标签的说明。
• the weights of all individual increments or units
所有单个份样或单元的重量
• If compositing is undertaken, the analytical sample weight is recorded
如果进行了混合操作,要记录分析样品的重量。
• notes on possible abnormalities or deviations
关于可能出现的异常情况或偏差的记录(说明)
• the name and signature of (all) the operator(s) involved.
相关操作人员(全部)的姓名及签名。
Second person review should ensure traceability and compliance throughout this phase.
应由第二人进行审核,以确保在整个这一阶段具有可追溯性且符合规定。
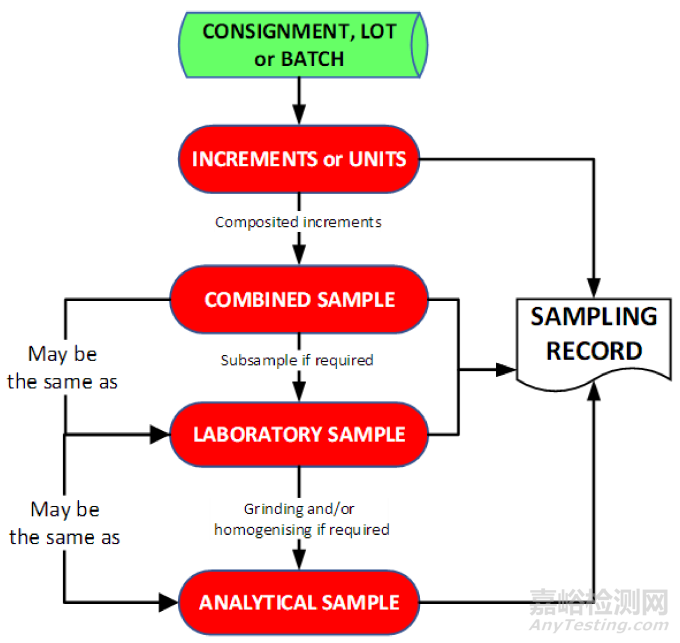
Figure 11 Proposed nomenclature and inputs to the sampling record
拟议的术语以及需录入取样记录的内容
9 Additional Guidance sources on sampling
关于取样的其他指导资料来源
Eurachem and the Royal Society of Chemistry have resources which provide information on sampling and, in particular, measurement uncertainty as listed below.
欧洲分析化学中心(Eurachem)和英国皇家化学学会(the Royal Society of Chemistry)拥有一些资源,这些资源提供了有关取样的信息,特别是关于测量不确定度的信息,如下所列。

10 Sampling is only part of the Analysis and Testing Process
取样只是分析和测试过程的一部分。
Whilst this guidance is concerned with sampling and sample management, it forms only the first, albeit critical, part of the journey. The testing aspect is covered in another guidance document, ECA_AQCG_SOP 03_APLM_v1.0_July 2018.
虽然本指南涉及取样和样品管理,但它只是整个流程的第一个部分(尽管是关键部分)。测试方面的内容在另一份指南文件《ECA_AQCG_SOP 03_APLM_v1.0_2018 年 7 月》中有所涵盖。
A high-level process flow is shown in Figure 11.
一个高层次的流程如图11所示。
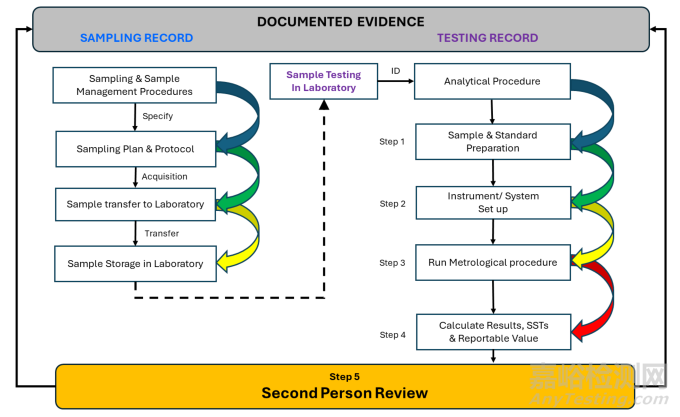
Figure 12 Traceability from sample to reportable value
从样品到可报告数值的可追溯性

来源:Internet
关键词: 实验室数据管理