
嘉峪检测网 2025-06-11 16:59
导读:文章旨在依托某大型医药公司已完成隔离器项目设计和验证策略,分享设计及系统验证实施过程中的关注点及注意事项,为后期隔离器的设计和验证提供参考。
隔离器作为疫苗生产行业中新兴设备,在解决生物安全暴露风险及无菌控制方面发挥着越来越重要的角色。实现有效的生物净化,内部环境与外部环境满足不受损害的、连续的隔离要求,注定隔离器在设计、制造、安装、调试与验证过程中区别于常规环境屏障。文章旨在依托某大型医药公司已完成隔离器项目设计和验证策略,分享设计及系统验证实施过程中的关注点及注意事项,为后期隔离器的设计和验证提供参考。
Part.01隔离系统的需求要点
1用户需求编制要点
质量源于设计,在用户需求中应明确隔离器舱体分段要求、空调机组系统、舱体焊接要求、门系统、控制系统、原位清洗/ 在线清洗系统、过氧化氢汽化灭菌系统、泄漏检测系统、过氧化氢在线浓度监测系统、温度检测系统、相对湿度检测系统、压差传感器、过滤器压差计、悬浮粒子计数器、浮游菌采样仪、手套袖套、配套支架、快速传递接口配置、风速监测、光栅系统、验证、售后等方面的规定。
2用户需求编制注意事项
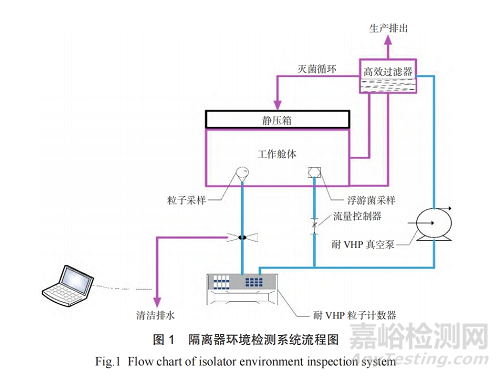
(1)根据美国注射剂协会第34 号技术报告要求[1],隔离器内部每立方米体积中需布置5~10 个生物指示卡用于隔离器汽化过氧化氢灭菌。生物净化灭菌工艺开发/ 灭菌工艺验证灭菌开发中需要大量生物指示卡、化学指示卡进行测试,费用成本较高,建议用户需求中明确指示剂的提供方式。(2)冻干、分装等存在粉尘暴露的产品,需提供第三方暴露限值验证报告,用户需求中需明确提供方式。(3)用户需求中应明确灌装机、轧盖机所有的真空管线(包括在线悬浮粒子及浮游菌)应能耐受过氧化氢蒸汽灭菌(完全达到杀灭106 的孢子效果,几乎无毒,可分解成水和氧气)[2]。所有真空泵的排气口需评估是否需经过袋进袋出过滤后排放到室外,如图1 所示。(4)欧盟无菌附录1[3]要求,隔离器手套完整性测试至少应在每个批次或阶段性生产的开始和结束时进行。隔离器配备手套较多,建议用户需求中根据设备手套数量确认配套手套测试仪的数量。
Part.02隔离器系统的验证要点
1木模测试
(1)设备供应商在签订合同后,根据人体尺寸数据,如亚洲人成年男子的平均肩高(站立)1494±5.6 mm, 肘高( 站 立 )1128±4.7 mm, 上臂长349 mm,前臂长268 mm ;亚洲人成年女子的平均肩高(站立)1385±5.6 mm,肘高(站立)1050±4.7 mm,上臂长319 mm,前臂长242 mm。搭建木条结构,以及木板通过铁钉的结合方式。依据设备平面布局图体现出操作腔体形貌、腔体内部的工作模块以及主要操作区域的整体高度。

(2)此过程需要企业与供应商多次进行现场的木模测试以保证隔离器定制的合理性、合规性。企业技术和关键操作人员,在木模现场模拟正常生产流程确认隔离器内手套孔位置和方向、快速传递接口主动阀位置、玻璃门位置和开门方向、在线浮游菌与悬浮粒子放置点、水枪位置、安全光栅位置、挂杆挂钩位置、排水口位置等。签订木模测试报告,开展下一步的设计确认工作。
2设计确认
新版GMP 附录1 中规定 :隔离操作器只有经过适当的确认后方可投入使用。确认时应当考虑隔离技术的所有关键因素,如隔离系统内部和外部所处环境的空气质量、隔离操作器的消毒、传递操作以及隔离系统的完整性[4]。
设计确认要点
根据功能说明、硬件设计说明、软件设计说明、报警清单、权限清单、电气图、管道仪表流程图、材料清单、木模确认结果,建立从用户需求到设计文件的追溯矩阵,举例如表1 所示。
表 1 用户需求与设计描述追溯矩阵示例表
Tab.1 Sample table of user requirements and design description traceability matrix
|
No.
|
URS No. | URS 描述 | 设计描述 | 设计参考 | 是否符合要求 | 备注 / 偏差报告编号 | |||
| FS | HDS | SDS | Other | ||||||
| 21 | URS19 | 隔离器各区之间采用鼠洞相连,鼠洞的设计应不影响各分区压差的建立。通过鼠洞的转盘或者网带应便于拆卸,且不影响相应位置的过氧化氢灭菌效果 | 隔离系统各舱体之间采用鼠洞门连接,鼠洞门不影响舱体内压差的建立,不影响网带安装,占用空间小,不影响生物净化效果。理瓶盘独立舱体,相对两边腔体为正压,与灌装段之间采用鼠洞设计 | Section 4.2 | N/A | N/A | P&ID | ☑ Yes 是 ☐ No 否 | N/A |
| 22 | URS20 | 该联动线的灌装机、孔盖机、外壁清洗机需配备手套,手套为进口品牌。可实现层流系统与灌装机的联动控制。需配备一个手套检测仪,同时能够满足至少 8 支设备两侧手套的同时监测 | 手套选用 Piercan 品牌,配备一拖八的手套检测仪 | Section 3 Section 4.2.2 | Section 10.1 | Section 8 | BOM (O) | ☑ Yes 是 ☐ No 否 | N/A |
设计确认注意事项
隔离器腔体和风管如需考虑灭活和清洗时,因喷头多、瞬时流量大,需考虑配置缓冲罐。各风管及各腔室清洗管路最远端具备排净功能,清洗的用量、流量、清洗控制方式等需要在设计确认中双方确认。
3工厂验收测试
隔离器在供应商工厂制作完毕后,开始进行工厂验收测试,主要内容如下 :管道制造资料审核、材质确认、抛光度确认、制造与设计符合性确认、整体布局图确认、管道仪表流程图确认、部件确认、电路图确认、硬件互锁确认、人机界面确认、输入/ 输出测试确认、安全访问确认、报表测试确认、界面功能及权限确认、审计追踪测试、配方开发/ 维护确认、报警确认、功能界面逻辑确认、断电和恢复确认、照明测试确认、噪声测试确认、门锁功能测试确认、舱体泄漏测试确认。

注意事项 :涉及到第三方配合的,如隔离器内部料液的转移方式需要在此阶段完成,对应的原位清洗、在线灭菌、功能配置硬件及程序控制方式需涉及第三方共同确认。
4现场确认
隔离器房间温度和湿度对于操作者的安全和舒适是重要的,温湿度对于除菌和净化技术的影响效果也很关键[5]。
现场验收确认要点
设备在现场安装调试后,进行现场验收测试,主要内容如下 :(1)设计符合性确认包括 :技术文件确认、关键部件的材料确认、测量元件合格证确认、仪器仪表校准确认、整体布局图确认、管道仪表流程图确认、高效过滤器完整性测试确认、部件确认、焊接文件符合确认、酸洗钝化符合确认、电路图确认、输入/ 输出测试确认、硬件控制确认、软件版本记录确认。(2)功能界面确认包括 :断电和恢复确认、界面功能确认、审计追踪测试、用户权限确认、配方开发/ 维护确认、报表测试确认。(3)运行确认包括 :报警功能确认、照明测试确认、噪声测试确认、压力控制测试确认、手套完整性测试、舱体泄漏测试确认、风速均匀性测试确认。(4)功能确认包括 :过氧化氢生物净化程序运行确认、清洗水排尽确认、气流流型确认、隔离器内洁净度检测确认(静态粒子计数)。
现场验收测试难点及注意事项
(1)清洗水排尽测试 :设备安装完毕后,需要对腔体坡度进行确认,用水枪向腔体内喷水,确保腔体内平面及排水口处无积水,避免后期清洗结束后出现积水。
(2)舱体泄漏测试 :泄漏检测是隔离器设备运行的重要环节,是能否进行生物净化的基础,也是费时最长的项目之一。隔离器内物品种类繁多,任何一个阀门、手套、卡盘接口、密封垫密封不严,都会导致泄漏率不合格。供应商多采用分段烟雾试验喷洒氨水,靠味觉和试纸监测的方式逐步查找漏点。
(3)灭菌工艺开发 :灭菌是指杀灭细菌及其繁殖体、芽孢、病毒和真菌孢子等一切形式的微生物的过程。国际上一般用“无菌保证水平”来评价灭菌效果,通常将无菌保证水平不大于百万分之一作为最终灭菌产品的无菌保证要求。过氧化氢因具有氧化还原作用而具有杀菌效果,过氧化氢灭菌技术,是利用过氧化氢在常温下气体状态比液体状态更具杀孢子能力的优点,通过复杂的化学反应解离具有强氧化性的羟基,来攻击细胞的成分,包括破坏细胞膜、脂类、蛋白质和DNA,达到完全灭菌要求的一种技术[6]。
隔离系统一般采用汽化的灭菌剂对内部环境进行表面灭菌,目前较常用的灭菌剂包括汽化过氧化氢、过氧乙酸等。灭菌剂发生器可集成于隔离系统中,也可独立于隔离系统,独立设计的灭菌系统与隔离系统之间的气体管路连接,应确保其密封性。灭菌剂应通过有效过滤后进人隔离系统内,灭菌结束后须对灭菌剂进行排空[7]。汽化过氧化氢灭菌作为隔离器现有的一种灭菌形式,对隔离器内无菌保障水平至关重要,灭菌的成功或失败,直接影响到产品的无菌性,产品的无菌性直接影响用药的安全。灭菌工艺的开发是保证灭菌工艺顺利实现必不可少的一环[8]。
1)灭菌工艺开发,主要是通过隔离器内部气流流型、温度分布、化学指示剂、生物指示剂、评估生产操作高风险区域等,确定出隔离器内净化时较差的点位及生产操作的高风险区域,通过过氧化氢汽化灭菌,使这些较差及高风险位置达到降低6 个对数值的要求,确定此时的灭菌参数。
2)灭菌工艺验证,通过灭菌工艺开发确定的灭菌参数、隔离器内装载的方式及较差及高风险位置进行化学指示剂和生物指示剂的布置,进行至少三次汽化过氧化氢灭菌,灭菌后所有指示剂变色或无菌生长作为验证合格的标准。
3)下文着重介绍项目灌装冻干隔离器灭菌开发的测试过程示例 :
①气流流型测试 :

通过气流流型测试,识别出隔离器内在汽化过氧化氢灭菌时气流的较差点位。按照确定的装载方式进行隔离器内物品的摆放及悬挂,使用灭菌参数及运行状态运行隔离器,用水雾发生器在接近隔离器均流膜处进行“发烟”,使水雾自上而下流下,并用摄像机记录每一次流型情况,直至隔离器全部拍摄完成。气流流型出现流型不佳、紊乱、流量较小等情况,即判定为气流流型较差点位置。为了方便隔离器内操作,气流流型拍摄时,灭菌时用注射用水替代过氧化氢。
②温度分布测试
温度对灭菌效果有着重要的影响,通过对隔离器内温度分布的研究,确认出温度较差位置。按照确定的装载方式进行隔离器内物品的摆放及悬挂,使用灭菌参数及运行状态运行隔离器,将温度探头布置在经过评估的位点上,监测灭菌周期内的温度情况,进而识别出温度与对比温度相差2 ℃和不符合24~32℃的区域,以此来判断温度分布的较差点位。本测试为了减少概率事件和结果的重现性,需要做3次或更多次测试以获得更加真实的数据。
③ 化学指示剂测试
通过化学的方法确认灭菌时汽化过氧化氢分布较差点位置,按照确定的装载方式进行隔离器内物品的摆放及悬挂,使用灭菌参数及运行状态运行隔离器,在运行灭菌前,将化学指示剂按照评估的位置进行布点,布点数量依据腔室内体积计算至少5 个/m3,布置时尽量能在隔离器外观察到指示条显色区,如果有视觉死角,可适当调整位置,灭菌结束后根据化学指示剂的结果确定出汽化过氧化氢分布较差点位置。
对比所有指示卡的变色时间及灭菌结束后变色的程度,识别出较差条件位置点。本测试为了减少概率事件和结果的重现性,需要做3 次或更多次测试以获得更加真实的数据。
④生物指示剂测试
通过生物的方法确认灭菌时汽化过氧化氢分布较差点位置。按照确定的装载方式进行隔离器内物品的摆放及悬挂,使用灭菌参数及运行状态运行隔离器,在运行灭菌前,把生物指示剂按照评估的位置进行布点,生物指示卡的菌含量大于106,菌种为嗜热脂肪芽孢杆菌,布点数量依据腔室内体积计算至少5个/m3,同时取未经灭菌的生物指示剂3 片同法接种,作为阳性对照[8]。将不含生物指示物的TSB培养基作为阴性对照物。重复上述微生物测试3 次。
灭菌程序结束后,取出测试样本,均放到TSB培养基中,嗜热脂肪芽孢杆菌芽孢在55~60 ℃条件培养7 天,测试微生物生长结果[9]。根据培养的结果确定出过氧化氢汽化灭菌分布较差位置。根据培养的结果调整灭菌参数,如参数有调整需要重新进行生物测试,直至灭菌后生物指示剂全部合格。本测试为了减少概率事件和结果的重现性,需要做3 次或更多次测试以获得更加真实的数据。
⑤生产操作高风险区域评估
根据隔离器内的工艺流程,评估出理瓶段、灌装加塞段、自动进出料段、轧盖段的生产操作的高风险区域,并作为汽化过氧化氢灭菌必须考虑的放置指示剂的点位,并在灭菌工艺验证过程中进行确认。
5安装确认/ 运行确认
安装确认/ 运行确认确认内容和注意事项与现场确认内容相同。
6性能确认
性能确认(PQ)是核实隔离器系统的功能是否符合操作者的要求。PQ 过程完成后,如果可能的话,灭菌周期的有效性和排出口的灭菌剂应该被检测。所有的PQ 过程的数据应该被统计,分析和存档[10]。
(1)性能确认前需确认手套完整性、舱体泄漏、生物净化等各项指标合格。建议按照“三静三动”策略对隔离器进行确认。静态测试可与洁净空调系统性能确认同步,动态测试建议在模拟操作过程(最差情况下的装载模式)、与模拟灌装同步(联动隔离器),验证生产最差挑战条件的无菌状态[9]。
(2)测试项目内容和标准要求同洁净空调系统,包括 :洁净度测试、沉降菌测试、浮游菌测试、表面微生物测试等。
(3)测试注意事项 :
① 隔离器可采取功能分区或者空调分区的原则划分功能段。验证点位的数量和位置需采用风险评估的形式确认,如网格法评估。若评估点位与在线悬浮粒子和浮游菌点位重合,可采用在线设施和数据。
② 生物净化前,验证用离线的浮游菌、悬浮粒子和培养基需清洁消毒后放置到隔离器内(无菌传递桶体积有限),设备无法耐受气化过氧化氢熏蒸部位可采用包裹形式保护。
③ 按照美国注射剂协会第34号技术报告[1]要求:隔离器中每0.5 m2 的底板面积悬浮粒子至少采样一次。风险评估中需结合此项规定。
④表面微生物要对无菌传递桶接口内表面、手套内表面、置物架(上方的置物杆及挂钩等)等部位选择取样。
7隔离器系统的再验证
为保障隔离系统在生命周期内的稳定运行,维持有效的验证状态,用户还应根据风险评估情况制定隔离系统的再验证计划。重要仪器仪表,例如压差仪表、温湿度仪表、风速仪表、流量仪表、粒子计数器、灭菌剂浓度传感器、称量天平等应定期进行校验。隔离系统的再验证一般包括年度验证和期间核查,用户应按照文件化的程序及规定的可接受标准实施再验证。再验证计划应围绕密闭系统的完整性,灭菌程序的有效性,无菌状态的维持能力等关键性能进行评估。再验证的结果应形成记录并保存。此外,用户在设备使用中,出现运行程序或参数变更、维护时更换重要配件、发生运行异常并完成维修后、安装场地变更以及长时间停用后的再启用等情况时,也应进行相应的再验证。
Part.03结论
隔离器作为一个“可移动的无菌室”,能最大限度地防止产品受到污染,为无菌灌装、无菌检验、高风险产品操作等提供了解决方案,在保护实验和生产人员安全中发挥了越来越重要的作用。本文通过对隔离器设备验证策略的开发与研究,进行了详细阐述和案例分析,总结了隔离器为了药品的无菌保证,在测试和验证阶段的关键内容,为隔离器验证实施提供了有力保证。
参考文献
[1] Food and Drug Admistraton. PDA Technical Report NO. 34.DESIGN AND VALIDATION OF ISOLATOR SYSTEMS FORTHE MANUFACTURING AND TESTING OF HEALTH CAREPRODUCTS【S】. Food and Drug Admistraton. USA. 2022:7-8,11-14.
[2] 刘万忠. 浅谈过氧化氢消毒技术的发展趋势[J]. 机电信息,2016. 16(29):41-44,49.
[3] European Commission. Volume 4 EU Guidelines for good quality control in the manufacture of medicinal products for human and veterinary use. Appendix 1 Production of sterile drugs [S].Brussels. 2022 :Section 4. 21
[4] 李晓雪,梁毅. 对无菌隔离操作器及其结构与确认的探讨[J].机电信息,2015,15(23):24-27,52.
[5] USP〈1208〉,Sterility Testing-Validation of lsolator System/General in formance[S]. 2020.
[6] 钱小进,陈敬华. 隔离器快速过氧化氢蒸汽灭菌程序开发及验证标准的建立[J]. 机电信息,2021,21(14):22-24.
[7] 国家药典委员会.《中国药典》(2020 年版)四部 :9206 无菌检查用隔离系统验证和应用指导原则[S]. 北京 :中国医药科技出版社,2020.
[8] 罗辉艳,严东珍,刘家媛,等. 无菌隔离器灭菌效果研究[J].中国设备工程,2017,33(6):78-79,82.
[9] 全国制药装备标准化技术委员会. 无菌隔离器 :JB/T 20175-2017[S]. 北京 :中国标准出版社,2017.
[10] 高海燕,丁恩峰,赵丽雅,等. USP 对无菌检验隔离器验证的技术要求[J]. 医药工程设计,2010,31(2):29-33.

来源:Internet
关键词: 隔离器