
嘉峪检测网 2025-06-30 08:59
导读:近日在对比审核新版ChP内容,特将两版《中国药典》1143细菌内毒素检查法进行对比。
细菌内毒检查法作为控制药品质量的一种常规、经济、有效方法,但是,在为新化合物或者新药建立细菌内毒素检查方法时,常常会遇到各种各样的困难,尤其是尚处在新药研发早期阶段的药物,由于药物制剂、赋性剂等不稳定给方法的建立带来不同程度的影响。
近日在对比审核新版ChP内容,特将两版《中国药典》1143细菌内毒素检查法进行对比:
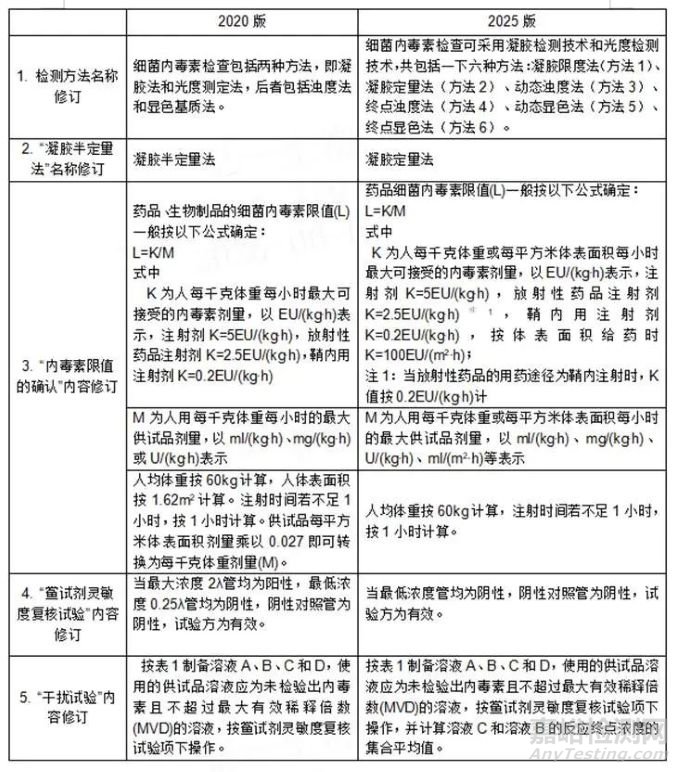
根据上表对比分析,2025版《中国药典》对细菌内毒素检测法的核心内容、操作要点等内容无修订,说明目前内毒素检测法对于现在市场或技术是满足的;在光度检测技术内容上无任何修改,也侧面说明目前该方法技术驱动不强或方法革新无任何推动。该次主要修订在内毒素限值、鲎试剂灵敏度复核试验有效性和细化检测方法等方面完善。因此该次2025版ChP修改对企业或者市场影响不大。
在药典中明确规定供试品检测时,可使用其中任何一种方法进行试验,当测定结果有争议时,除另有规定外,以凝胶限度试验结果为准。在此仅从一种新药(X注射剂)的细菌内毒素方法建立及其检测角色出发,在此浅谈细菌内毒素检查法-凝胶限度法。
原理:通过鲎试剂与内毒素产生凝集反应的原理进行限度检测或定量检测内毒素的方法。
方法建立工作需要系统化完成以下关键步骤,确保方法的科学性、合规性、可重复性。
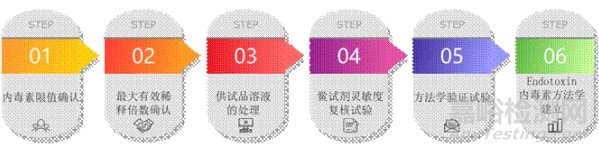
1.内毒素限值(L)的确认:一个药品在投放市场前,它是否满足内毒素限值要求?样品内毒素含量具体是多少?这些问题不但关注用药安全,还最大限度的提供有关内毒素含量的准确信息,为药品生产中质量控制提供警戒信息,尽管内毒素检查法的建立是一个科学方法的研究过程,但其最终目的还是要为控制药品质量服务,因此,在方法建立时,不但要阐明限值的合理性,考虑技术可能达到的限值,同时还要满足相关药品管理法规的要求。
内毒素限值L的公式计算:L=K/M,举例其中已知X注射剂的K=5EU/(kg.h),X注射剂的人体最大用量为每小时1.2g,按照我国人均体重60Kg计算,M=1.2g/h ÷60Kg=0.02g/kg.h=20mg/kg.h,即X注射剂内毒素限值L=K/M=0.25EU/mg,假定药品X的浓度为100mg/ml,那么内毒素限值L=0.25EU/mg ×100mg/ml=25EU/ml。
2. 确定最大有效稀释倍数(MVD):MVD是指在所选择的鲎试剂灵敏度条件下,对产品进行最大倍数的稀释,仍能够进行内毒素限值的检测,此时所允许稀释的最大倍数。计算公式:MVD=内毒素限值(L)×药品浓度(C)÷鲎试剂灵敏度(λ),鲎试剂有多种规格,比如0.03EU/ml、0.06EU/ml、0.25EU/ml。根据上步确认L为0.25EU/mg,假定药品X的浓度为10mg/ml,当选择0.25EU/ml的鲎试剂,即MVD=10;当选择0.03EU/ml的鲎试剂,MVD=83。
最小有效浓度(MVC)是指在所选择的鲎试剂灵敏度条件下,将产品稀释到一定浓度,仍能够进行内毒素限值的检测,此时所允许的最小浓度。MVC=鲎试剂灵敏度(λ)/内毒素限值(L)。
对选定的鲎试剂灵敏度来讲,MVC时一个绝对值,MVD对特定的药品浓度时特异的,浓度变化时,MVD也会随之发生变化。
3. 供试品溶液的制备:确认药品基本信息:药品的分子量大小、规格、浓度、体积等,以便选择合适的药品处理方法和内毒素检测方法。如果在处理过程中运用了激烈的处理条件或溶剂,还应证明采用方法不会破坏或者减少药品中细菌内毒素含量,并且排除药品pH、盐浓度(推荐等渗环境)、温度(一般不超过40℃)或β-葡聚糖等因素的干扰。
如何排除pH的干扰呢?假设药品X的pH为3.90,一般而言供试品溶液和鲎试剂混合后溶液的pH值在6.0-8.0的范围内最为适宜。我们可以使用Tris缓冲液作为稀释液,通过1M氢氧化钠溶液来调节药品的pH。当然1M氢氧化钠和Tris缓冲液应该在已去除内毒素的容器中配制,且不含内毒素和干扰因子。
4. 鲎试剂灵敏度复核试验:选择并确认鲎试剂使用规格,按照药典或者厂家说明书要求制备细菌内毒标准品,然后制成2λ、λ、0.5λ、0.25λ 4个浓度的内毒素标准液,取不同浓度的内毒素标准液分别与等体积的鲎试剂溶液(T)混合,每个内毒素浓度平行做4管,另取2管加入等体积的细菌内毒素检查用水(BET)作阴性对照,根据下表进行加样处理。将试管中溶液轻轻混匀后,封闭管口,垂直放入37℃±1℃的恒温器中,保温60分钟±2分钟。
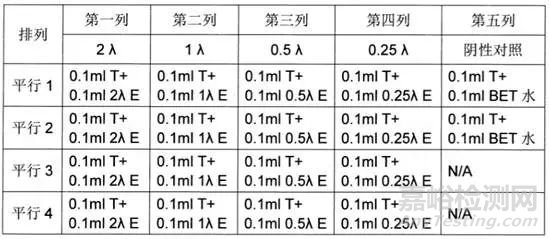
判断方法 将试管从恒温器中轻轻取出,缓缓倒转180°,若管内形成凝胶,并且凝胶不变形、不从管壁滑脱为阳性;未形成凝胶或形成的凝胶不坚实、变形并从管壁滑脱为阴性。
接受标准 当最低浓度管均为阴性,阴性对照管为阴性,试验方为有效。按下式计算反应终点浓度的集合平均值,即为鲎试剂灵敏度的测定值(λc),
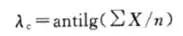
式中:X为反应终点浓度(指系列递减的内毒素浓度中最后一个呈阳性结果的浓度)的对照值(lg);n为每个浓度的平行管;
当λc在0.5λ-2λ(包括0.5λ、2λ)。方可用于细菌内毒素检查,并以标示灵敏度为该批次鲎试剂的灵敏度。
5. 方法学验证试验:
1) 预干扰实验:预干扰试验目的是初步确认最小不干扰的药品稀释浓度(NIC),为正式干扰试验提供适当药品稀释浓度依据。预干扰试验对一系列药品X的稀释液进行试验,在每一个稀释浓度都有一组添加内毒素的供试品溶液和一组不添加内毒素的供试品溶液,供试品浓度可逐级稀释,但最终供试品的稀释倍数不超过MVD。根据下表制备。
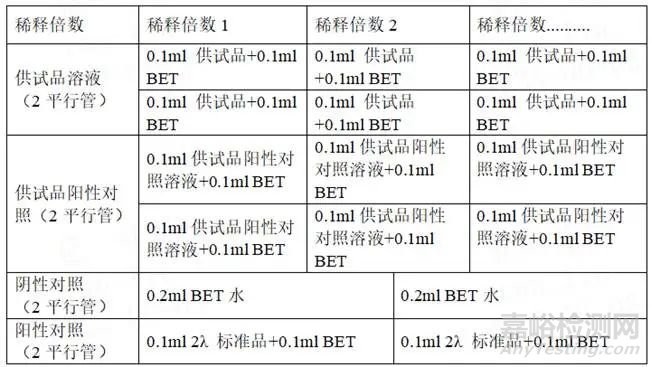
2) 干扰试验:按照下表制备A、B、C、D,使用的供试品溶液应为未检验出内毒素且不超过最大有效稀释倍数(MVD)的溶液。
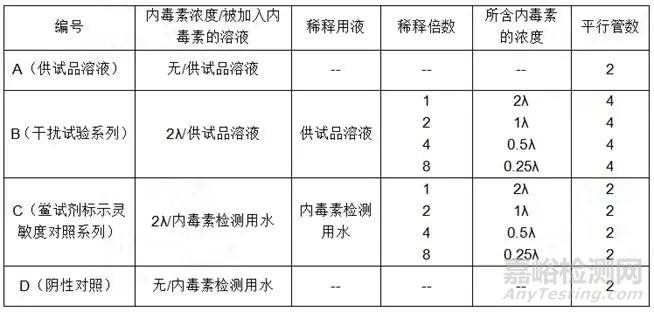
接受标准 只有当溶液A和阴性对照溶液D的所有平行管均为阴性,并且系列溶液C的结果复核鲎试剂灵敏度复核试验要求时,试验方有效。
结果判断 当系列溶液B的结果复核鲎试剂灵敏度复核试验要求时,认为在该浓度下的供试品溶液无干扰作用。其他情况则认为供试品在该浓度下存在干扰作用。若供试品溶液在小于MVD的稀释倍数下对试验有干扰,应将供试品溶液进行不超过MVD的进一步稀释,再重复干扰试验。
6. 凝胶限度试验方法确认:撰写方法学验证试验报告,并且对药品X的细菌内毒素检查完成日常检测与质控的SOP,其内容一般包含样品处理步骤、鲎试剂复溶方法、孵育时间、温度控制、结果判断标准等。具体加样方式可参照下表来制备溶液A、B、C、D。其中使用稀释倍数不超过MVD并且已经排除干扰的供试品溶液。
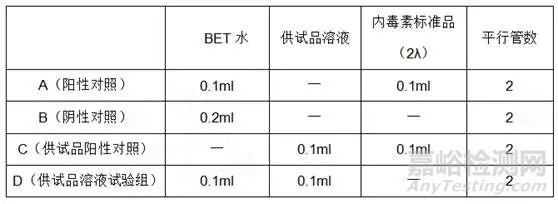
结果判断 在37℃±1℃恒温器中保温60分钟±2分钟后观察其结果(判断方法与鲎试剂灵敏度复核试验一致)。若阴性对照均为阴性,阳性对照管和供试品阳性对照管均为阳性,试验方有效。若供试品两个平行管均为阴性,则判断供试品合格。若供试品两个平行管均为阳性,则判定不符合规定。若供试品两个平行管一管为阳性、一管为阴性,则需要进行复试。复试时供试品溶液C需做4支平行管,若所有平行管均为阴性,则判定供试品符合规定,否则判定不符合规定。
参考文件:
1. 2020版《中国药典》
2. 2025版《中国药典》
3. [医药]细菌内毒素检查方法的建立、限值的确定 - 豆丁网

来源:药事纵横
关键词: 细菌内毒素检测