
嘉峪检测网 2025-07-11 20:59
导读:lnavolisib片剂区分力溶出方法和接受标准。
|
产品信息 |
|
|
NDA序号 |
219249 |
|
规格 |
3 mg和9 mg |
|
使用途径 |
口服 |
|
申请人 |
GENENTECH, Inc. |
|
治疗分类 |
肿瘤 |
|
RLD |
新分子实体 |
|
拟定适应症 |
磷脂酰肌醇-4,5-二磷酸3-激酶(PIK) 催化亚基α亚型蛋白(p110α;由PIKCA基因编码)的选择性抑制剂,用于治疗乳腺癌 |
|
产品外观 |
薄膜衣片: • 3 毫克:红色圆形凸面片剂,一面刻有"INA 3"字样。 • 9 毫克:粉色椭圆形片剂,一面刻有"INA 9"字样。 |
原料药信息:
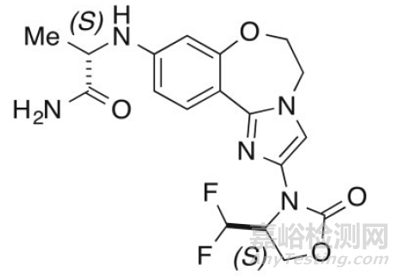
商品名含有(lnavolisib, 一种激酶抑制剂。Inavolisib的化学名为(2S)-2-[[2-[(4S)-4-(二氟甲基)-2-氧代-噁唑烷酮-3-基]-5,6-二氢咪唑并[1,2-d][1,4]苯并氧氮䓬-9-基]氨基]丙酰胺。Inavolisib为白色至类白色、灰粉色、灰橙色或灰黄色粉末。Inavolisib的分子式为C18H19F2N5O4,分子量为407.37 g/mol。其化学结构如下所示:
TRADENAME薄膜包衣片有两种规格可供口服,分别含有3毫克和9毫克的inavolisib。本品还含有乳糖、硬脂酸镁、微晶纤维素。

贮存在20°C至25°C(68°F至77°F)条件下,允许在15°C至30°C(59°F至86°F)之间短暂波动[参见USP控制室温要求]。
NDA 219249 是针对新分子实体(NME)的原研1类新药申请,旨在批准lnavolisib片剂(3 mg和9 mg)作为速释制剂。该药物活性成分是磷脂酰肌醇-4,5-二磷酸3-激酶(PIK)催化亚基α亚型蛋白(p110α;由PIK3CA基因编码)的高效选择性抑制剂,拟用于乳腺癌患者的治疗。
拟商业化制剂为速释型(b)(4)薄膜衣片(FCT),口服给药,提供3 mg和9 mg两种规格。剂量规格设计旨在满足精准便捷的给药方案:使用9 mg片剂单次给药9 mg,或通过3 mg片剂实现剂量递减至6 mg或3 mg。
本次生物药剂学审评重点包括:
1) 对拟用受试制剂溶出度检测方法及可接受标准的评估与适用性判定;
2) 对制剂桥接必要性的评估。
体外溶出度方法和接受标准:
根据申请人提供的全部信息和数据,以下溶出度方法和接受标准被认为是充分且可接受的。
在临床开发过程中处方的变更桥接:申请人开发了标记为F01-F17的一系列拟定处方药品,贯穿整个临床开发阶段。申请人通过体外溶出度研究桥接了临床研究期间进行的各种处方变更。从早期处方(F01)到II期及主要稳定性处方(F19),片剂在pH 1和pH 6.8条件下均显示出与速释产品药物释放行为一致的相似溶出特性。
申请人提出的溶出方法及检测条件(3mg规格使用500 mL 0.1N HCl、9mg规格使用900 mL介质,采用USP第二法(桨法),50转/分钟,37±5°C),两规格批次放行和稳定性测试的溶出接受标准均为Q=80%(30分钟内),该方案对lnavolisib片剂具有充分适用性。
生物药剂学总体建议
从生物药剂学的角度来看,建议批准新药申请NDA-219249-ORIG-1所申报的Inavolisib片剂(3毫克与9毫克规格)。
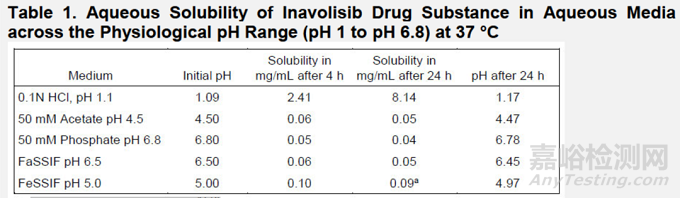
溶解度:如表1所示,原料药inavolisib在pH依赖性(b)(4)溶解度
渗透性:申请人认为Inavolisib的被动渗透性为中等(MDCK细胞试验表观渗透系数1.9×10^-6厘米/秒;流速渗透系数0.9×10^4厘米/秒)。
溶出度:拟定的Inavolisib片剂(3 mg和9 mg)在采用USP溶出度测定法II、桨速50 rpm、溶出介质为500 mL 0.1 N盐酸且不含表面活性剂的条件下,显示快速溶出特性(b)(4)。基于(b)(4)的考量,溶出介质体积确定为500 mL 0.1 N盐酸。
溶出方法与接受标准:
背景:
Inavolisib被归类为一种具有高水溶性且中等渗透性的分子。其制剂设计为速释片剂。单次和多次给药后均显示剂量-暴露量呈线性关系。据申请人数据,单次口服9 mg[14C]标记inavolisib胶囊后的绝对生物利用度为76%(90%置信区间:69-83%),表明该药物具有较高的绝对生物利用度(临床总结模块2.7.1)。由于拟定制剂具有快速溶出特性(30分钟内药物释放(4)%),预计药物释放速率对整体吸收速率的影响极小。
溶出度方法:
Inavolisib的溶解度数据(表2)表明该化合物具有pH依赖性溶解特性,根据生物药剂学分类系统可归类为高溶解性分子。基于此评估,申请人参照2018年FDA工业指南《含有高溶解性药物的速释固体口服制剂溶出度测试及验收标准》,采用(b)(4)条件下的溶出测试方案用于制剂生产及有效期内的常规质量控制。由于Inavolisib两种规格片剂(3mg与9mg)具有剂量比例性且采用相同生产工艺(b)(4),初始阶段应用了相同的体外溶出方法和测试条件。申请人提出的溶出度方法与验收标准详见表2。

根据2024年6月3日IR回复,使用900 mL溶出介质用于9 mg规格产品已被验证合理性,并被判定为可接受。
解读:两种上市规格,小规格3mg采用了500ml介质体积、大规格9mg采用了900ml介质体积的溶出方法进行放行,pH均为0.1N HCl,说明化合物在水性介质中溶解度较低,采用了较强酸性介质。体内数据也表明,药物体外的释放速率对其整体吸收速率影响较小。
溶出方法区分能力:
申请人评估了所拟定的溶出度测试条件对单位处方故意变化的区分能力,并证明原料药粒度分布(PSD)对溶出度没有影响。图9A展示了分别采用D10为(b)(4)微米、D50为(b)(4)微米、D90为(b)(4)微米的原料药细颗粒、标准及粗颗粒处方制备的3 mg片剂的溶出曲线。图9B展示了分别采用D10为(b)(4)微米、D50为(b)(4)毫米、D90为(b)(4)毫米的原料药标准及粗颗粒配方制备的9 mg片剂的溶出曲线。
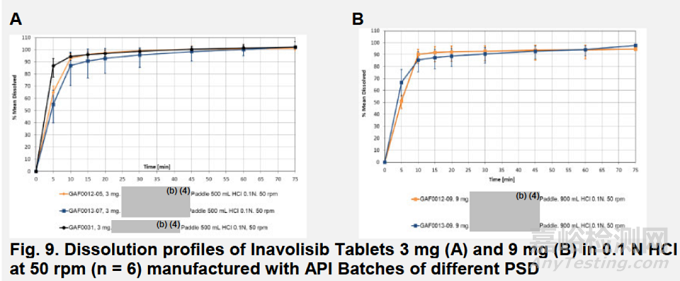
申请人还研究了片剂硬度对溶出度(b)(4)的影响。(b)(4)溶出数据(图10A和B)表明,所提出的溶出方法能够区分不同硬度制备的片剂。
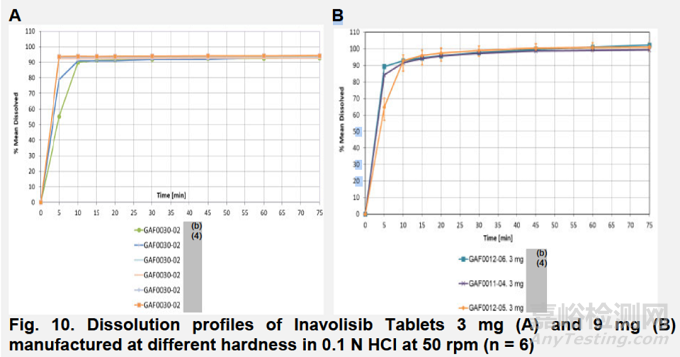
除了PSD和片剂硬度外,申请人还研究并证明了所提出的溶出方法在不同储存和强力条件(不同温度和湿度)下的区分能力。
评估:根据申请人提供的整体信息,审评人员认为该药物制剂拟定的溶出度测试条件(3mg规格采用500 mL 0.1N HCl介质、9mg规格采用900 mL 0.1N HCl介质,USP第二法装置,50转/分钟,37#5°C)经以下支持性信息验证具有可接受性:
1. 拟定产品为速释剂型,旨在胃滞留时间及pH范围内实现快速药物释放。
2. 根据BCS分类系统,活性成分Inavolisib在胃液pH范围内属于高溶解性药物。
3. 被确定为关键物料属性(CMA)的API粒度分布对药物释放无显著影响。原料药粒度已得到充分控制(b)(4)。
4. 拟定溶出方法对片剂硬度、贮存条件及强力条件具有区分力。
5. 由于Inavolisib在生理pH条件下具有高溶解性,被确定为制剂关键质量属性(CQA)的溶出度具有极低的初始生物药剂学风险,因此对拟定药物的生物利用度影响可忽略。
6. 拟定溶出方法及测试条件(USP第二法装置,50转/分钟,0.1N HCl介质500 mL/900 mL)符合2018年《溶出度指导原则》对含高溶解性药物制剂的要求,且符合2021年《M9基于生物药剂学分类系统的生物等效性豁免指导原则》相关规定。
7. 溶出度接受标准Q=30分钟内释放(%)(b)(4)具有合理性。
8. 溶出度标准可确保药物完全释放(b)(4)。
解读:化合物溶解度呈pH依赖性,Inavolisib制剂属速释制剂,在生理pH条件下可快速释放,因此其对药物的生物利用度影响风险极低。将产品体外释放的快慢与体内生物利用度区分开,释放速度对生物用利度的影响很小。
开发的溶出方法介质虽采用了较强酸性介质,仍对压片硬度和强力条件下的样品有适度的区分力,对原料药粒度没有区分力,说明溶出方法可以较好的区分不同条件的产品,表现较好的质量控制能力。

来源:蒲公英OurYao
关键词: lnavolisib片剂 区分力 溶出方法 接受标准