
嘉峪检测网 2025-07-15 10:13
导读:药品上市后,经常会遇到原料药发生变更情形,原料药变更相关指导原则有《已上市化学药品药学变更研究技术指导原则(试行)》和《已上市化学药品药学变更研究技术指导原则(试行)》原料药变更的问答。本篇结合上述指导原则,将原料药变更对制剂厂家的影响进行梳理,以供理解记忆。
药品上市后,经常会遇到原料药发生变更情形,原料药变更相关指导原则有《已上市化学药品药学变更研究技术指导原则(试行)》和《已上市化学药品药学变更研究技术指导原则(试行)》原料药变更的问答。本篇结合上述指导原则,将原料药变更对制剂厂家的影响进行梳理,以供理解记忆。
1、思维导图
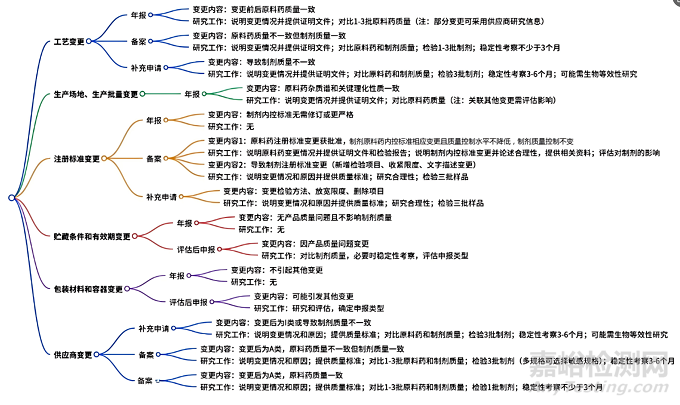
2、变更详情
2.1 生产工艺变更
|
变更级别 |
变更内容 |
研究工作 |
|
年报 |
变更前后原料药的质量(如杂质谱、关键理化性质等)一致 |
(1)简要说明原料药的变更情况,提供相关证明性文件(如批准证明性文件、备案通过的证明性文件等)。 (2)对变更前后 1-3 批原料药进行质量对比研究,原料药的杂质谱、关键理化性质(如晶型、粒度等)等应保持一致。 (注:如变更原料药的过程控制、起始原料内控标准、中间体内控标准等,且经评估对制剂质量的潜在影响较小,上述研究工作也可采用原料药供应商提供的研究信息。) |
|
备案 |
变更前后原料药的质量(如杂质谱、关键理化性质等)不一致,但变更前后制剂的质量(如溶出曲线、杂质谱、关键理化性质等)保持一致时 |
(1)简要说明原料药的变更情况,提供相关证明性文件。 (2)对变更前后 1-3 批原料药进行质量对比研究。 (3)对采用变更前和变更后原料药制备的制剂进行全面的质量对比研究,变更前后制剂的溶出曲线、杂质谱、关键理化性质等应保持一致,并符合相关指导原则的要求。 (4)对采用变更后原料药连续生产的 1-3 批制剂进行检验。 (5)对采用变更后原料药生产的 1 批制剂进行加速及长期稳定性考察,提供不少于3个月的稳定性研究资料,并与变更前产品的稳定性情况进行比较,变更后产品的稳定性不低于变更前。 |
|
补充申请 |
原料药的变更导致相关制剂的质量(如溶出曲线、杂质谱、关键理化性质等)不一致 |
(1)简要说明原料药的变更情况,提供相关证明性文件。 (2)对变更前后的原料药进行质量对比研究。 (3)对采用变更前和变更后原料药制备的制剂进行全面的质量对比研究,应符合相关指导原则的要求。 (4)对采用变更后多批原料药连续生产的 3 批制剂进行检验。 (5)对采用变更后原料药生产的 3 批制剂进行加速及长期稳定性考察,提供 3-6 个月的稳定性研究资料,并与变更前产品的稳定性情况进行比较,变更后产品的稳定性不低于变更前。 (6)如变更前后制剂的溶出曲线及可能影响生物利用度的关键质量属性等不一致,一般需考虑进行生物等效性研究,如申请免除生物等效性研究,需进行充分的研究和分析。 |
2.2 生产场地、生产批量变更
|
变更级别 |
变更内容 |
研究工作 |
|
年报 |
原料药的杂质谱和关键理化性质等应保持一致 |
1、简要说明原料药的变更情况,提供相关证明性文件。 2、对变更前后的原料药进行质量对比研究,变更前后原 料药的杂质谱、关键理化性质等应保持一致。 (注:如变更原料药的生产场地、变更生产批量的同时还关联其他变更(如工艺参数、生产设备变更等),制剂持有人应评估变更对原料药和制剂质量的影响,参考工艺变更进一步确定制剂的变更类别及相关研究工作。) |
2.3 注册标准变更
|
变更级别 |
变更内容 |
研究工作 |
|
|
年报 |
经评估制剂中原料药的内控标准及制剂的注册标准无需修订,或制剂仅为制定更严格的原料药内控标准(如收紧限度、增加检验项目、变更文字描述等) |
/ |
|
|
备案 |
当原料药的注册标准变更已经获得批准,制剂持有人经评估,制剂的原料药内控标准的项目(如已批准的原料药注册标准的变更项目)如需进行相应变更,且变更后原料药的质量控制水平不降低,也不影响制剂的生产和质量控制 |
(1)说明原料药注册标准的变更情况,提供相关证明性文件及原料药供应商的检验报告。 (2)说明制剂的原料药内控标准的变更情况,论述变更的合理性,提供变更前后的原料药内控标准和检验报告,如分析方法进行了变更,还需提供相关的方法学验证资料,变更后原料药的质量控制水平不得降低。 (3)评估原料药内控标准的变更对制剂的影响,变更后不影响制剂的生产和质量控制。 |
|
|
备案 |
原料药注册标准的变更导致制剂的注册标准变更 |
(1)新增检验项目。 (2)在原标准规定范围内收紧限度。 (3)注册标准中文字描述的变更,此类变更不应涉及检验方法、 限度等的变更。
|
(1)说明具体变更情况和原因,提供变更后的质量标准。 (2)对质量标准变更合理性进行研究。 若涉及限度修订,需对一定批次样品(建议含近效期样品)批分析结果进行汇总,为限度修订提供依据。另外,需考察在原定的有效期内,药品是否符合修订后质量标准的要求。 如涉及增加检验项目,需对检验方法进行方法学研究(包括方法的选择、验证)、提供限度拟定依据。需对一定批次样品(建议含近效期样品)批分析结果进行汇总,以考察在原定的有效期内,药品是否符合修订后质量标准的要求。 (3)按变更后的质量标准对三批样品进行检验,应符合规定。 |
|
补充申请 |
(1)变更检验方法。 (2)放宽控制限度。 (3)删除注册标准中的任何项目。 |
(1)说明具体变更情况和原因,提供变更后的质量标准。 (2)对质量标准变更合理性进行研究。 如涉及检验方法改变,需对新方法进行方法学研究验证并应与变更前方法进行比较,确保方法变更不引起药品质量控制水平的降低。 另外,需对一定批次样品(建议含近效期样品)批分析结果进行汇总,以考察在原定的有效期内,药品是否符合修订后质量标准的要求。 如涉及删除标准中任何内容,需结合药品生产过程控制、药品研发过程及药品性质等综合分析和证明该项变更不会引起药品质量控制水平的降低。 如涉及放宽控制限度,需进行详实的研究,必要时需要有关安全性和/或有效性试验资料或文献资料的支持。限度变更还需基于一定批次样品的检测数据并符合相关的技术指导原则。 (3)按变更后的质量标准对三批样品进行检验,应符合规定。 |
|
2.4 贮藏条件和有效期变更
|
变更级别 |
变更内容 |
研究工作 |
|
年报 |
如原料药不存在产品质量问题,且经制剂持有人评估认为原料药的变更不太可能影响制剂的质量,制剂持有人可在年报中报告。 |
/ |
|
评估后申报 |
如因产品质量问题(如原批准的贮藏条件下和/或有效期内产品不符合质量标准的要求等),原料药变更了贮藏条件和有效期 |
建议制剂持有人对变更前后的制剂进行全面的质量对比研究,必要时进行稳定性考察,结合研究结果评估是否进行年报、备案或提出补充申请。 |
2.5 包装材料和容器变更
|
变更级别 |
变更内容 |
研究工作 |
|
年报 |
当原料药的包装材料和容器发生变更时,不引起其他变更 |
/ |
|
评估后申报 |
如该变更可能引发其他的变更 |
制剂持有人应进行相应的研究和评估,并根据评估结果进行年报、备案或补充申请。 |
2.6 供应商变更
|
变更级别 |
变更内容 |
研究工作 |
|
补充申请 |
变更后的原料药为I,或者当原料药供应商的变更导致相关制剂的质量(如溶出曲线、杂质谱、关键理化性质等)不一致 |
(1)说明变更的具体情况和原因。 (2)提供变更前后原料药的质量标准。 (3)对变更前后的原料药进行全面的质量对比研究。 (4)对采用变更前和变更后原料药制备的制剂进行全 面的质量对比研究,应符合相关指导原则的要求。 (5)对采用变更后多批原料药连续生产的3批制剂进行检验,应符合相关指导原则的要求。 (6)对采用变更后原料药生产的3批制剂进行加速及长期稳定性考察,申请时提供3-6个月的稳定性研究资料,并与变更前产品的稳定性情况进行比较,变更后产品的稳定性不低于变更前。 (7)如变更前后制剂的溶出曲线、可能影响生物利用度的关键质量属性等不一致,一般需考虑进行生物等效性研究,如申请免除生物等效性研究,需进行充分的研究和分析。 |
|
备案 |
变更后的原料药为A,且当变更前后原料药的质量(如杂质谱、关键理化性质等)不一致,但变更前后制剂的质量(如溶出曲线、杂质谱、关键理化性质等)保持一致时 |
(1)说明变更的具体情况和原因。 (2)提供变更前后原料药的质量标准。 (3)对变更前后 1-3 批原料药进行全面的质量对比研究。 (4)对采用变更前和变更后原料药制备的制剂进行质量对比研究,变更前后制剂的溶出曲线、杂质谱、关键理化性质应保持一致,并符合相关指导原则的要求。 (5)对采用变更后原料药连续生产的 3 批制剂进行检验,应符合质量标准的规定。对于多规格且处方工艺相同的制剂,可选择对原料药的变更最敏感的规格进行研究,其余规格可适当减少研究批次,一般来说对低剂量药物使用最低规格,其它药物使用最高规格,应提供充分的选择依据。复杂剂型因风险相对较大,不建议减少批次。 (6)对采用变更后原料药生产的3 批制剂进行加速及长期稳定性考察,申请时提供3-6 个月的稳定性研究资料,并与变更前产品的稳定性情况进行比较,变更后产品的稳定性不低于变更前。 |
|
备案 |
变更后的原料药为A,且当变更前后原料药的质量(如杂质谱、关键理化性质等)保持一致时 |
(1)说明变更的具体情况和原因。 (2)提供变更前后原料药的质量标准。 (3)对变更前后1-3 批原料药进行全面的质量对比研究,原料药的杂质谱、关键理化性质(如晶型、粒度等)等应保持一致。 (4)对采用变更前和变更后原料药制备的1-3批制剂进行质量对比研究,变更前后制剂的溶出曲线、杂质谱、关键理化性质应保持一致,并符合相关指导原则的要求。 (5)对采用变更后原料药生产的1 批制剂进行检验,应符合质量标准的规定。 (6)对采用变更后原料药生产的1 批制剂进行加速及长期稳定性考察,申请时提供不少于3 个月的稳定性研究资料,并与变更前产品的稳定性情况进行比较,变更后产品的稳定性不低于变更前。 |

来源:Internet