
嘉峪检测网 2025-08-23 16:15
导读:本文首次完整归纳了国内外9个药品监管机构、国际组织及行业协会等发布的历版数据完整性规范、指南、技术报告和问答等文件的时间表,详细分析了各自的重点内容,相关文件已基本涵盖了药品实验室主要分析领域。同时,对目前仍容易产生分歧的数据完整性概念理解和工作做法进行了明确。
摘 要 / Abstract
目的:系统性掌握药品实验室数据完整性领域的国内外监管机构和行业的相关要求和主要做法。方法:全面梳理国内外9 个药品监管机构、国际组织及行业协会等发布的数据完整性规范、指南、技术报告和问答,归纳各自的主要特点和应用方向,并对历史版本的演变内容进行介绍。对数据完整性的重要概念和主要做法结合工作实际提出解读意见,对数据完整性最新的监管理念进行解读,对信息化条件下的药品实验室数据完整性方面的实际问题进行分析并给出建议。结果:本文首次完整归纳了国内外9个药品监管机构、国际组织及行业协会等发布的历版数据完整性规范、指南、技术报告和问答等文件的时间表,详细分析了各自的重点内容,相关文件已基本涵盖了药品实验室主要分析领域。同时,对目前仍容易产生分歧的数据完整性概念理解和工作做法进行了明确。此外,结合工作经验提出了目前药品实验室审计追踪的注意事项、实验室数据采集满足数据完整性属性的注意事项、实验室数据归档注意事项、实验室中动态数据和混合系统数据的保存注意点、实验室数据管理软件主要验证考虑事项等方面的工作建议。结论:本文通过对数据完整性相关文件发展演变进行系统性阐述,提出了解决药品实验室数据完整性关键问题的思路,以期为构建数据完整性合规的药品实验室提供有益参考。
Objective: This study aims to systematically examine the requirements and practices of domestic and international regulatory agencies and industry stakeholders in ensuring data integrity within pharmaceutical laboratories. Methods: A comprehensive review was conducted of data integrity standards, guidance, technical reports, and Q&A documents published by domestic and international drug regulatory authorities, international organizations, and industry associations. Key characteristics and areas of application were summarized, along with a historical overview of their evolution. Core concepts and best practices were interpreted in light of practical experience, and the latest regulatory perspectives on data integrity were analyzed. Common challenges faced under increasingly digital laboratory conditions were explored, with recommendations proposed. Results: This paper is the first to comprehensively compile a timeline of key data integrity documents published by nine major regulatory and professional bodies. It provides a detailed analysis of their main focus areas, covering essential aspects of laboratory analysis. At the same time, clarification is provided on frequently misunderstood concepts divergent practices. In addition, based on field experience, the paper presents practical recommendations for audit trail practices, data collection strategies that align with data integrity principles, data archiving, handling of dynamic and hybrid data systems, and software validation considerations. Conclusion: This paper provides a systematic explanation of the evolution of data integrity requirements, offering insights into addressing key issues related to data integrity in drug laboratories. It serves as a valuable reference for establishing compliantand robust data integrity management systems in pharmaceutical laboratories.
关 键 词 / Key words
数据完整性;药品监管机构;行业协会;规范;指南
data integrity; drug regulatory authority; industry association; regulatory standards; guidance
数据完整性(data integrity)是药品全生命周期管理的重要组成部分,是药品质量与安全的重要保障。在国内外药品监管机构对药品生产企业的合规检查中,数据完整性问题是主要问题之一,其中实验室环节发生的数据完整性问题在药品生产企业数据完整性问题中占主要部分[1-4]。随着药品实验室信息化进程加速,计算机化系统[5]、电子实验笔记本[6]等工具广泛应用,对数据完整性的管理要求已从传统纸质记录延伸至电子数据全生命周期。目前,全球多个药品监管机构均将数据完整性作为重点监管领域,并相继发布多份行业规范和实施指南等文件[7-8]。在确保数据完整性要求的前提下,这些文件的倾向和侧重点均存在差异,甚至部分观点也存在差异。例如,部分文件侧重于数据完整性的总体要求和概念解释,有些则侧重于提供药品实验室具体检查使用指导和微生物专门实验室指导;部分文件仅涉及良好生产质量管理规范(good manufacturing practice,GMP)、良好供应和管理规范(good distribution practice,GDP)领域,有些则涵盖GxP( 包括GMP、GDP、GCP 等)领域;部分文件强调对具体数据的细节要求,有些则强调从全生命周期以及数据完整性文化的角度进行考量。此外,部分文件在数据审计追踪方面的要求也有所不同,并且随着版本的更新,其监管思路亦呈现动态演进特征。现有研究已对数据完整性领域相关行业规范和指南等展开解读[8-11],但仍存在完整的各版本历史演变解读不全、行业规范和指南等文件之间的综合理解和解读较少、药品实验室各分析领域侧重点的解读缺乏、数据完整性要求的最新发展解读不足等问题。基于此,笔者从药品实验室角度出发,对比分析了国内外9 个药品监管机构、国际组织及行业协会等发布的数据完整性规范和指南等文件的主要特点,并对目前药品实验室存在的数据完整性关键问题进行了初步探讨,以期通过系统性阐述数据完整性发展演变历程,精准把握相关文件的核心要义。同时,结合笔者工作经验,针对药品实验室在数据完整性方面存在的主要问题提出解决思路,从而为构建符合数据完整性合规要求的药品实验室提供有益参考。
01数据完整性的历史和定义
1.1 数据完整性监管的历史沿革
数据完整性问题并非近年才凸显的监管议题。早在1963 年,美国食品药品监督管理局(Food and Drug Administration,FDA)就为了解决数据完整性问题,发布了《美国联邦法规》(Code of Federal Regulations,CFR)第21 篇第133 条款(以下简称21CFR 133),该条款在1975 年变更为21 CFR 210《制造、加工、包装或者保存药品的优良制造规范》(Current Good Manufacturing Practice in Manufacturing, Processing, Packing, or Holding of Drugs; General) 和21 CFR 211《动态药品生产管理规范》(Current Good Manufacturing Practice for Finished Pharmaceuticals) 两部分。1997 年8 月20 日,FDA正式发布21 CFR 11《电子记录与电子签名》(Electronic Records; Electronic Signatures),这是首个针对电子数据管理的法规。值得注意的是,21 CFR 11 中未直接定义“数据完整性”这一术语,而是以“记录完整性”的概念来替代。
1.2 数据完整性与数据可靠性
数据完整性和数据可靠性是data integrity 的不同中文译法。2016 年10 月原国家食品药品监督管理总局发布了《药品数据管理规范(征求意见稿)》(2016 年版)[12],首次提出“数据可靠性(Data Integrity)” 并对其进行定义:数据可靠性是指贯穿整个数据生命周期的数据采集是完整的、一致的和准确的程度。所收集的数据应该是可归属的,清晰的,同步记录的,原始的或真实副本,并且是准确的。2017 年8月发布的《药品数据管理规范(征求意见稿)》(2017 年版)[13],对数据可靠性的定义进行了完善:数据可靠性指在数据生命周期内,数据完整、一致、准确的程度。应当以安全的方式收集和维护数据,从而保证数据归属至人、清晰可溯、同步记录、原始一致、准确真实(国际上,常用缩略词“ALCOA”或“ALCOA+”概括)。
数据完整性的定义最早见于英国药品和健康产品管理局(Medicines and Healthcare Products Regulatory Agency,MHRA)2015 年3 月发布的《GMP 数据完整性定义和行业指南》(GMP Data Integrity Definitions and Guidance for Industry)[14], 其中指出数据完整性(data integrity) 是所有数据在整个数据生命周期中的全面、一致和准确的程度。MHRA在2 0 1 8 年发布的《GxP 数据完整性指南和定义》(‘GXP’ Data Integrity Guidance and Definitions)[15] 中指出, 数据完整性是数据的完整、一致、准确、可信和可靠程度,以及数据生命周期中数据这些属性得到维护的程度。数据应以安全方式收集和保存,从而保障数据归属至人、清晰可溯、同步记录、原始(或真实副本)和准确。世界卫生组织(World Health Organization,WHO) 在2016 年6 月发布的《良好数据和记录管理实践指南》(Guidance on Good Data and Record Management Practices)[16]中指出,数据完整性是指数据完整、一致、准确、可信和可靠的程度, 以及在整个数据生命周期中保持这些数据特征的程度。数据应以安全的方式收集和保存, 从而保障数据归属至人、清晰可溯、同步记录、原始或真实副本及准确。综合来看,MHRA 和WHO 均在相关文件中指出,数据完整性要求数据具有归属至人(attributable)、清晰可溯(legible)、同步记录(contemporaneous)、原始一致(original) 和准确真实(accurate)的属性(即ALCOA),以及完整(complete)、一致(consistent)、持久(enduring)和可及(available)的属性(即ALCOA+)[15,17]。
考虑到国家药品监督管理局(National Medical Products Administration,NMPA)于2020 年7 月发布的《药品记录与数据管理要求(试行)》[18] 中已经不再使用数据可靠性一词,同时结合国内外相关行业对不同译法的接受程度,本文暂采用数据完整性进行表述。
02国内外数据完整性要求特点及相关文件
笔者收集并梳理了1997 年以来国内外9 个药品监管机构、国际组织及行业协会等发布的关于数据完整性的关键性规范、指南、技术文件等,详细信息见表1。
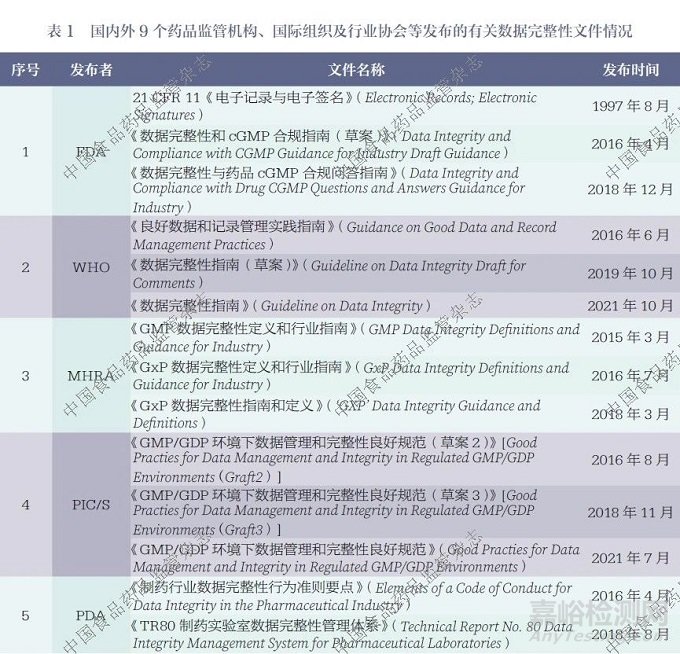

2.1 美国食品药品监督管理局
目前FDA 尚未专门针对数据完整性问题发布完整且独立的指南文件,但在其相关法规要求和行业指南等文件中,已有部分内容涉及数据完整性管理的原则性规定。2018 年12 月,FDA 发布了《数据完整性与药品 cGMP合规问答指南》(Data Integrity and Compliance with Drug CGMP Questions and Answers Guidance for Industry)[19],该指南阐述了FDA 对于制药企业在数据完整性方面重点关注且容易存在缺陷的18 个问题的态度,特别是对一些共性问题进行了详细论述,包括关于计算机系统公用登录账户问题,单机计算机设备保存纸质打印件或静态记录而非原始记录的问题, 在“系统适用性”测试或试检、预检、平衡运行中使用实际样品的问题,以及仅保留重新处理的实验室色谱最终结果而未保留原始数据的问题等。FDA 认为,数据完整性问题属于21 CFR210、21 CFR 211 和21 CFR212 所规定的《现行良好生产规范》(Current Good Manufacturing Practice,cGMP) 基本问题。ALCOA 原则的所有重要属性在cGMP 中均有相应的监管依据,并且FDA 针对每个问题都给出了cGMP 条款参考。基于此,FDA认为21 CFR 11、21 CFR 210、21 CFR 211 和21 CFR 212 已对制药企业的数据完整性提出了实质性要求,因此没有必要再发布专门的指南文件。
2.2 英国药品和健康产品管理局
2015 年3 月,MHRA 发布《GMP 数据完整性定义和行业指南》(GMP Data Integrity Definitions and Guidance for Industry)[14], 该指南介绍了MHRA 对于数据完整性的立场,明确了达成合规要求的最低期望,并提出了基于风险的数据管理方法,包括数据风险、数据关键程度和数据生命周期等方面。这是首个涵盖了数据完整性相关术语定义和要求的指南,为正确理解数据完整性问题提供了具有积极意义的参考。此后,MHRA分别在2016 年7 月和2018年3 月发布了《GxP 数据完整性定义和行业指南》(GxP Data Integrity Definitions and Guidance for Industry)[20] 和《GxP 数据完整性指南和定义》[15]。在这2 份指南文件中, 数据完整性的适用范围从制药行业扩展至GxP 领域,但不包括医疗器械和药物警戒领域。同时, 指南文件明确指出, 其主要涉及数据完整性,而不是数据质量,ALOCA+ 中的“+” 是为了强调相关要求而添加的,所以无论是ALCOA 还是ALCOA+, 其在数据完整性的期望方面并无差异。由于英国是经济合作与发展组织(Organization for Economic Co-operation and Development,OECD) 的成员国,因此在良好实验室管理规范(good laboratory practice,GLP)领域,OECD 发布的《良好实验室规范工作组关于GLP数据完整性咨询文件》(Advisory Document of the Working Party on Good Laboratory Practice on GLP Data Integrity)[21] 具有更高的优先级,其效力优于MHRA 的相关指南。
2.3 欧洲药品管理局
与FDA 类似, 欧洲药品管理局(European Medicines Agency,EMA)也未发布专门的数据完整性指南,而是采用问答的形式,阐述了其对数据完整性的监管理念。2016 年8 月,EMA 在“良好生产规范和良好销售规范指南: 问题和回答”栏目中,增加了对数据完整性方面23 个问题的回复内容。EMA 指出,要通过国际人用药品注册技术协调会(ICH)发布的《Q9 :质量风险管理》(Q9: Quality Risk Management) 中的方法, 对数据全生命周期的各个阶段开展数据风险性和数据关键性评估,并在此基础上,采用相应的控制措施来实现最终的数据完整性目标。EMA 同时强调了数据完整性的监管期望是数据需要符合ALCOA原则,并提供了每项ALCOA 原则与欧盟GMP 参考条款的对应关系,表明欧盟GMP 条款能够满足数据完整性的管理要求。
2.4 中国国家药品监督管理局
2016 年10 月和2017 年8 月先后发布的《药品数据管理规范(征求意见稿)》的2 个版本,提出了数据可靠性的概念并进行了修订。在这2 个版本中,规范文件的适用范围从“药品研制、生产、流通等活动,包括从事上述活动的临床试验、合同研究(CRO)、委托生产(CMO)、委托检验等单位和个人”,修订为“药品研发、生产、流通、上市后监测与评价等产品生命周期中全部活动的数据管理”。同时,数据的管理原则从“数据管理应贯穿整个数据生命周期,坚持真实、准确、及时、可追溯的数据管理原则,确保数据可靠性(Data Integrity)”,修订为“数据管理应当遵守归属至人、清晰可溯、同步记录、原始一致、准确真实的基本要求,确保数据可靠性”,并对数据各项ALCOA 属性进行了相应的管理规定,强调了数据的全生命周期管理理念。不过,该规范的正式版本至今尚未发布。
NMPA 在2020 年7 月发布了《药品记录与数据管理要求(试行)》[18], 该要求自2020 年12月1 日起施行。其中明确指出,其适用范围为在我国境内从事药品研制、生产、经营、使用活动中产生的,应当向药品监督管理部门提供的记录与数据。同时,该要求对记录和数据的概念进行了清晰界定,并分别针对纸质记录和电子记录制定了相应的规定,这些规定主要倾向于原则性的管理要求。
2.5 世界卫生组织
WHO 在2016 年6 月发布了《良好数据和记录管理实践指南》,此后,该指南历经更新,于2019 年10 月和2021 年10 月分别被《数据完整性指南(草案)》(Guideline on Data Integrity Draft for Comments)[17] 和《数据完整性指南》(Guideline on Data Integrity)[22] 所取代。WHO 的指南较早地对数据完整性进行了定义,但随着版本不断更新和升级,至《数据完整性指南》发布时,已不再对数据完整性进行定义解释,而只保留了对数据完整性ALCOA+ 属性要求的相关内容, 这体现了其指导思想的转变。除了强调数据全生命周期管理和质量风险管理的重要性之外,WHO 还特别强调了管理层在数据完整性方面应承担的职责和企业质量文化所产生的影响,同时提出了对纸质数据的优良文件规范要求以及对计算机化系统完整性的要求等。尤其值得关注的是,指南附录1 中列举了11 个数据完整性管理案例,这些案例对正文中的数据完整性要求起到了辅助说明的作用。同时,针对纸质数据、电子数据和混合系统数据的各项ALCOA 属性要求,案例也均进行了具体细化,具有较强的指导意义。
2.6 药品检查合作计划
药品检查合作计划(Pharmaceutical Inspection Co-operation Scheme,PIC/S)是一个由各国(地区)药品监管机构在GMP 领域组成的非正式组织。该组织的主要职责是制定GMP 领域的通用标准,并为检查员提供相关培训机会,因此其工作重点侧重于GMP 检查员角度。PIC/S 分别于2016 年8 月、2018 年11 月和2021 年7 月发布了《GMP/GDP 环境下数据管理和完整性良好规范》(Good Practies for Data Management and Integrity in Regulated GMP/GDP Environments)的第2 稿草案[23]、第3 稿草案[24] 以及定稿指南[25],版本的升级主要是在计算机化系统方面完善了数据传输和混合系统的完整性相关要求。PIC/S 强调数据要符合ALCOA+原则,并且列举了ALCOA+ 各原则与该指南条款的对应关系。从数据全生命周期的范围出发,PIC/S 基于质量风险管理角度,对纸质系统、计算机化系统和混合系统的数据完整性提出了具体要求,并且从检查的视角提出了各环节未达到预期时应关注的风险点。此外,PIC/S 还强调了质量文化在保障数据完整性方面发挥的重要作用,并且提出了对外包活动的数据完整性要求。同时,PIC/S 对数据完整性缺陷进行了分类,并给出了解决数据完整性问题的具体方法流程。
2.7 美国注射剂协会
美国注射剂协会(Parenteral Drug Association,PDA) 在2016 年4 月发布的《制药行业数据完整性行为准则要点》(Elements of a Code of Conduct for Data Integrity in the Pharmaceutical Industry)[26],对制药行业数据完整性的关键点进行了概述,例如,数据收集、分析、报告和保存的要求,电子数据采集系统要求,员工培训要求,数据完整性问题的上报流程以及对问题行为的调查等。PDA 在2018 年8 月发布了《TR80 制药实验室数据完整性管理体系》(Technical Report No. 80 Data Integrity Management System for Pharmaceutical Laboratories)[27]技术报告,该报告提出了药品监管机构对数据完整性的要求和期望,并将人为因素作为质量控制(quality control,QC) 实验室数据完整性问题的考虑要素之一。同时,该报告还系统论述了微生物实验室和QC 实验室数据完整性的管理要求,特别是考虑到微生物检测以手工操作为主的数据完整性特点,分析了实验室混合系统、计算机化系统和紫外- 可见分光光度仪、红外分光光度仪等其他仪器设备的数据完整性要求,以及实验室数据管理软件在数据完整性方面的要求。此外,PDA 还建议将ICH Q9 的质量风险管理理念应用于数据治理中,并提出了对数据完整性方面的漏洞进行纠正的方法。该报告为药品实验室在数据完整性管理方面提供了具体且实用的操作方法。
2.8 国际制药工程协会
国际制药工程协会( International Society for Pharmaceutical Engineering,ISPE) 在2016 年6 月发布了《电子记录与数据完整性指南(草案)》(Electronic Records and Data Integrity Graft), 并于2017年3 月正式发布了《记录与数据完整性指南》(Records and Data Integrity Guide)[28]。ISPE 强调,数据应符合ALCOA+ 原则,对于数据完整性的管理应是基于ICH Q9 质量风险管理理念的数据全生命周期治理。该指南指出了数据治理需要充分考虑人为因素,并且在涵盖管理、发展、操作等内容的14 个附录中提供了具体的数据完整性操作指导。值得注意的是,ISPE 还引入了数据完整性成熟度评级方法。该方法基于文化、治理与组织、战略规划和数据完整性计划、法规、数据生命周期、数据生命周期支持过程等6 个方面共36 个评价因素,对企业的数据可靠性进行5 个等级评价,等级从低到高依次为一级至五级。这一评估方式能够帮助企业找到自身在数据完整性管理方面存在的不足,并为其指明提升数据完整性水平的最佳方向。
2.9 欧洲原料药委员会
欧洲原料药委员会(Active Pharmaceutical Ingredients Committee,APIC) 分别于2019 年3 月和2022 年4 月发布了《基于风险的数据完整性管理实践指南》(Practical Risk-Based Guide for Managing Data Integrity)的第1 版[29] 和第2 版[30]。相较于第1 版,第2 版指南主要新增了审计追踪管理相关内容,补充了中级和中高级严重性数据来源和例子,并将示例重新命名为附件,移至独立的附件文档中。该指南着重介绍了一种全面的数据完整性风险管理方法,具体而言,它依据数据全生命周期的特点对数据进行分类管理,明确各类数据对应的系统最低要求;同时,系统性地评估数据管理中各体系和流程存在的差距,进而开展风险评估,并根据风险的严重程度制定相应的风险管理措施。此外,该指南采用业务流程图和工作表的形式,提供了一系列实用的数据完整性风险管理工具。2023 年4 月,APIC 发布了一份数据完整性常见问题的技术资料,针对数字和电子签名、密码管理、访问管理以及记录的生命周期管理等4 个方面的11 个相关问题给出了指导意见。
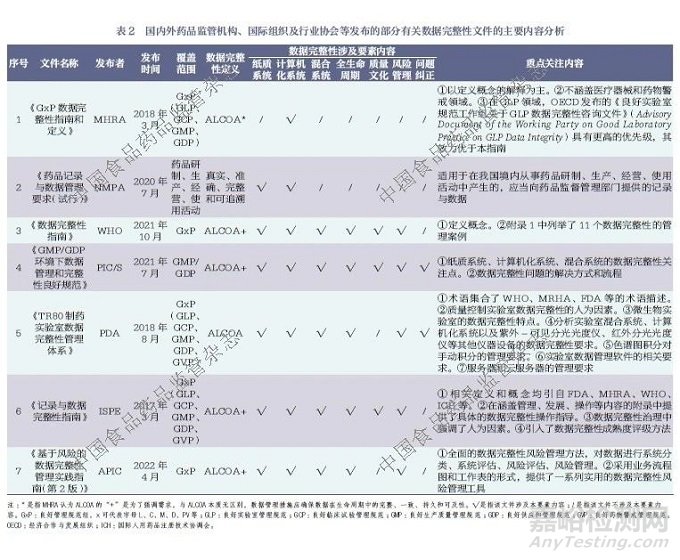
对于上述发布的数据完整性规范、指南、技术报告等文件,笔者对部分文件的涵盖范围、数据完整性属性、数据完整性涉及要素内容以及重点关注内容进行了统计分析,详见表2。
03数据完整性相关重要概念及关系
3.1 数据与记录的关系
数据与记录有着密不可分的关系。NMPA 发布的《药品记录与数据管理要求(试行)》中指出,“数据是指在药品研制、生产、经营、使用活动中产生的反映活动执行情况的信息,包括:文字、数值、符号、影像、音频、图片、图谱、条码等;记录是指在上述活动中通过一个或多个数据记载形成的,反映相关活动执行过程与结果的凭证。”由此可见,数据是药品相关活动执行过程中产生的信息,而记录是反映药品相关活动执行情况的凭证。简言之,记录是数据的凭证化呈现,数据是记录所承载的信息内容,两者在实质内容上是一致的,只是在载体形式上有所区别。
3.2 完整性数据的属性特征
国内外药品监管机构、国际组织及行业协会等对数据完整性的认识基本一致, 普遍认为数据应该具备ALCOA 属性, 甚至需达到ALCOA+ 属性标准。ALCOA 属性的具体特征为:可追溯的(attributable), 即能够追溯到数据生成的具体个人,并在必要时追溯到测量系统;可读的(legible),即数据和元数据在数据的整个生命周期内应可读;同步的(contemporaneous),即相关人员应在数据和信息生成或获取时立即进行记录;原始的(original),即原始记录(或经验证的真实副本),是首次获取的数据或信息,以及为全面重建GxP 活动实施过程所需的所有后续数据,均应可获取;准确的(accurate), 即数据是正确的、真实的、完整的、有效的和可靠的。ALCOA+ 属性是在ALCOA 属性的基础上,进一步增加了CCEA 特征,具体含义为:完整的(complete), 即数据必须是一个完整的集合;一致的(consistent),即数据必须相互一致;持久的(enduring),即数据是耐用的,并且贯穿整个数据生命周期;可获取的(available),即数据随时可用于审查或检查目的。ALCOA+ 属性主要是从数据使用需求的角度,进一步强化了对数据完整性的要求,但其本质上与ALCOA 属性没有差别,都是为了确保数据的质量和可靠性。
3.3 数据保存、备份、归档、传输和迁移
在数据完整性的相关规范和指南等文件中,对于数据保存、备份、归档、传输和迁移这几个名词的解释基本类似。数据保存是以归档(针对需要长期存储而受保护的数据)或备份(为应对灾难恢复而保存的数据)为目的的数据处理过程。数据备份是在规定的时间间隔内,以安全的方式复制实时电子数据,以确保在需要时数据能够被成功恢复。数据归档是在规定的保存期限内,对记录进行长期存储和保护的过程,目的是防止其变质、被篡改或删除。此外, 归档还包括将电子记录从活动数据库中删除。数据传输是在不同的数据存储类型、格式或计算机化系统之间传输数据的过程。数据迁移是将已存储的数据从一个持久的存储位置转移至另一个存储位置的过程。
数据的保存、备份、归档、传输和迁移都需要经过严格验证,以确保其满足数据完整性的要求。具体而言,数据备份是短期行为,旨在应对突发情况下的数据恢复;而数据归档是长期行为,侧重于数据的长期保存和保护。PDA、ISPE 和PIC/S 指出,备份数据不应就地存档,而是应存于距离较远(物理分离)的位置。对于数据传输,PIC/S 发布的《GMP/GDP 环境下数据管理和完整性良好规范》中特别指出,数据传输要确保数据直接传输到安全的位置和(或)数据库,而不是简单地从本地驱动器复制,这是因为本地复制存在数据被篡改的风险。数据迁移过程中可更改数据的格式,但不应更改内容或含义,且迁移后原存储地址不再保留原始数据。
3.4 混合系统数据管理要点
混合系统是指一种集数据管理和控制功能于一体的系统,通常由生成电子数据的电子系统和生成纸质记录的人工系统组成。因此,来自混合系统的完整数据组会同时包括电子数据和纸质数据。ISPE 发布的《记录与数据完整性指南》以及PIC/S 发布的《GMP/GDP 环境下数据管理和完整性良好规范》均明确指出纸质流程和电子流程之间的接口是混合系统数据完整性的风险点,强调应建立相应的程序并做好记录,以控制手动系统和自动系统之间的接口,尤其是人工生成的数据手动录入计算机系统、将自动化系统生成的数据转录(包括手动转录)到纸质记录上,以及自动检测打印数据并将其转录到计算机化系统中等。此外,还需要注意的是,纸质原始记录经过扫描生成电子副本后,尽管该副本可被认定为真实副本,但对于纸质记录的处置仍需谨慎,必须遵循相关法律法规、监管要求以及机构内部管理制度等的要求,来决定纸质记录是否可以被销毁,并严格执行必要的管理程序。
3.5 对数据完整性的理解
结合国内外药品监管机构、国际组织及行业协会等对于数据完整性要求的理解,实现数据完整性就是要在数据的全生命周期内,建立一套基于风险评估的完善的数据治理体系,包括良好的文件管理规范以及数据完整性文化氛围,以此确保数据始终具备ALCOA+ 属性特征。数据完整性工作不存在唯一的标准做法,而是需要与各自的实际工作相结合。只有深入理解并合理运用上述概念, 尤其是准确把握ALCOA+的内涵,才能真正达到数据完整性的目标。
3.6 审计追踪的范围
审计追踪作为元数据的一种表现形式,包含了与GxP 记录的创建、修改或删除等操作相关的信息。审计追踪是实现数据完整性的基本要求,无论是纸质系统还是电子系统,均需满足这一要求;在具备条件的计算机化系统中,其更是必要条件。对于计算机化系统中审计追踪范围的理解,MHRA 在其发布的《GxP 数据完整性指南和定义》中指出,审计追踪不必要包括每个系统活动,如用户登录或退出、敲键等操作无需记录在审计追踪中;FDA 发布的21 CFR 11 第11.10 (e) 节要求,使用安全的、计算机生成的、带有时间戳的审计追踪,独立记录操作员输入的日期和时间,以及创建、修改或删除电子记录的操作;ISPE 发布的《记录与数据完整性指南》进一步阐释,FDA这一要求特别针对操作员的输入和操作,包括创建、修改或删除受监管的电子记录,但不包括用户执行的所有活动,也不包括所有系统操作;APIC 发布的《基于风险的数据完整性管理实践指南》中提出,应对关键数据进行审计追踪记录,其中关键数据指的是那些可能影响产品质量的数据。从上述描述来看,审计追踪主要是针对影响关键数据的信息进行追踪,并非对全部行为活动的信息都进行追踪。
3.7 质量文化因素对数据完整性的重要性
当前,实验室质量文化因素对数据完整性的重要性正日益受到关注。WHO 在《数据完整性指南》中指出,高级管理层有责任营造一个有利于建立、保持和持续改进质量文化的环境,并且支持透明和公开地报告组织各级偏差、错误或遗漏和数据完整性失误等情况。PIC/S 发布的《GMP/GDP 环境下数据管理和完整性良好规范》中提出,质量文化是管理层、团队负责人、质量人员以及所有有助于创建质量文化以确保数据质量和完整性的人员,所共同展示出的价值观、信念、思维方式和行为模式的集合。ISPE 在《记录与数据完整性指南》中强调,人为因素在数据完整性中有着关键性影响。人为因素涵盖多个方面,包括公司和当地的文化、事故类型及原因、人为失误、数据伪造和欺诈、不公正压力以及适当的行为控制等。其中,公司和当地的文化影响是需重点考虑的因素之一。综合上述描述可以看出,在实验室中,构建自上而下全员参与的数据完整性质量文化体系,强化全员质量意识,并实施行之有效的数据完整性质量行为和质量机制,是确保数据完整性的关键要素。
04药品实验室数据完整性管理相关思考
针对药品实验室如何有效落实数据完整性相关要求这一问题,笔者根据国内外数据完整性相关规范和指南等文件,以药品监管机构、国际组织及行业协会等提出的相关要求为基础,对其中需要重点关注的事项进行了归纳,并提出如下思考。
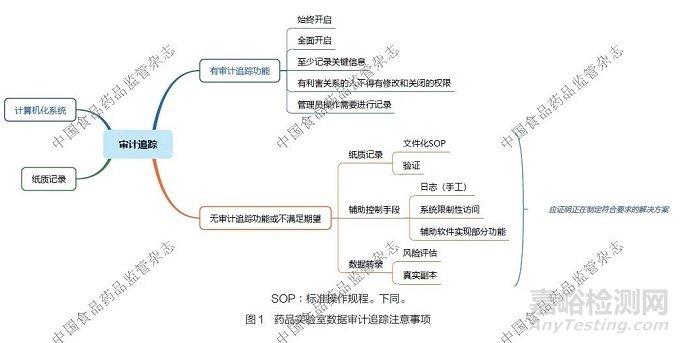
4.1 药品实验室数据审计追踪的注意事项
对于实验室中具备审计追踪功能的计算机化系统,应全面且始终开启审计追踪功能,同时至少记录关键操作信息。与数据有利害关系的人员不得有修改和关闭审计追踪功能的权限,即使是信息技术管理员进行操作,也应进行详细记录。对于实验室中不具备审计追踪功能的计算机化系统,需采取有效的辅助控制手段,如建立日志(含手工记录日志)、实施系统限制性访问措施、借助辅助软件实现部分审计追踪功能等。对于实验室的纸质记录,应制定文件化的操作规范,并进行相关的操作验证,以确保记录过程的规范性和准确性。对于无审计追踪功能的计算机化系统所涉及的数据转录操作,应该先进行数据完整性风险评估,并且要确保转录的数据为原始数据的真实副本。实验室应将所有无审计追踪功能或不满足相关期望的系统纳入改进计划,制定符合要求的解决方案并推进实施。药品实验室数据审计追踪的注意事项详见图1。
4.2 药品实验室数据采集满足ALCOA+ 属性的注意事项
药品实验室数据采集满足ALCOA+ 属性要求是保障数据完整性的根本。针对不同类型的实验数据,笔者分别阐述了满足相关要求的注意事项,详见图2。
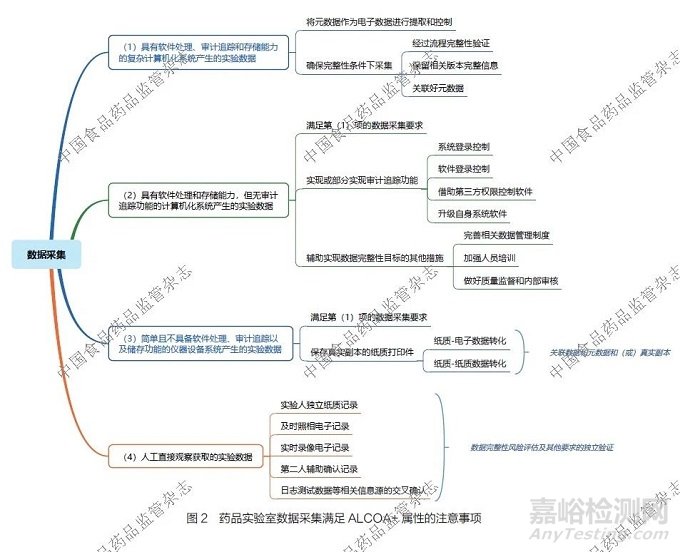
(1)具有软件处理、审计追踪和存储能力的复杂计算机化系统产生的实验数据。对于此类实验数据,如液相色谱仪、气相色谱仪以及各类质谱仪等产生的数据:①应尽最大努力将元数据作为电子数据进行提取和控制。②若数据以图片、PDF 等固定电子格式形式采集,在经过流程完整性验证、保留相关版本完整信息,以及关联好元数据的前提下,可认可其具备数据完整性。
(2)具有软件处理和存储能力,但无审计追踪功能的计算机化系统产生的实验数据。对于此类实验数据,如紫外- 可见分光光度仪、红外分光光度仪、电位滴定仪等产生的实验数据:①要尽量满足第(1)项下关于数据的采集要求。②要通过系统登录控制、软件登录控制,借助第三方权限控制软件,以及升级自身系统软件等措施,实现或部分实现审计追踪功能,至少要实现关键权限控制功能。③完善相关数据管理制度,加强人员培训,做好质量监督和内部审核等工作,也可以辅助实现药品实验室数据完整性的目标。
(3)简单且不具备软件处理、审计追踪以及储存功能的仪器设备系统产生的实验数据。对于此类实验数据, 如pH 计、天平、不溶性微粒分析仪等产生的实验数据:①要尽量满足第(1)项下关于数据的采集要求。②若仪器设备具备同步数据打印功能,且经过验证打印数据是数据和元数据的真实副本时,可保存打印数据。由于部分仪器采用的是热敏纸,需要采用经过验证的程序进行纸质- 电子数据转化或者纸质-纸质数据转化,以实现长期保存,并尽量与元数据和(或)真实副本有效关联。
(4)人工直接观察获取的实验数据。对于此类实验数据,如显色、沉淀、产气、显微、比色和薄层等产生的实验数据,在经过数据完整性风险评估及其他要求的独立验证后,可根据风险和要求的不同,采用以下1 种或多种数据采集形式:实验人独立纸质记录、及时照相电子记录、实时录像电子记录、第二人辅助确认记录,以及日志测试数据等相关信息源的交叉确认等。
4.3 药品实验室数据归档注意事项
药品实验室在进行数据归档时,须遵循以下3 个原则:①对等原则。无论是电子数据还是非电子数据,均应同等重视并予以规范处理。②对象原则。归档的数据必须是原始记录和(或)经过验证的真实副本。③可用原则。归档的数据要在数据有效期内具备可检索和可读取等特性。药品实验室数据归档注意事项详见图3。
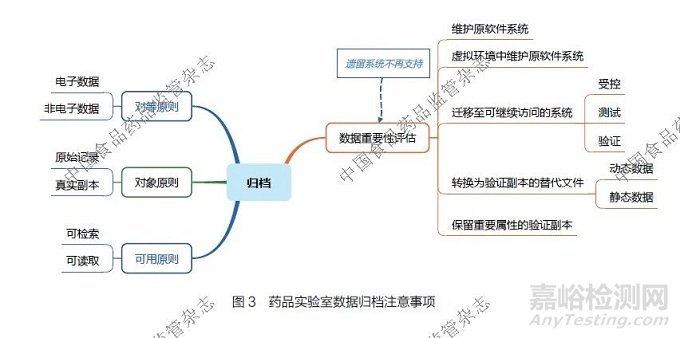
若实验室数据系统进行升级,可能会出现遗留系统无法再支持数据读取的情况。此时,可以根据对数据重要性的评估结果,制定分级的数据归档策略:①应优先考虑尽量维护原软件系统。②若不能在原软件系统中继续维护,可以尝试将原软件系统迁移至虚拟环境中继续进行维护。③若在虚拟环境中维护也不可行,则需将数据迁移至一个可继续访问的系统。该系统应处于受控状态,且经过测试和验证后,证明其能实现相应功能。④若实在无法找到合适的系统来维护数据,可将原始数据转换为经过验证的真实副本的替代文件(包含动态数据和静态数据)进行归档。⑤作为最后手段,可对数据中的重要属性部分进行验证副本的转化,然后进行归档。
4.4 药品实验室中动态数据和混合系统数据的保存注意点
实验室中动态数据和混合系统数据的保存应遵守ALCOA+原则、根据预定用途原则和保存原始数据和元数据原则,其注意事项详见图4。
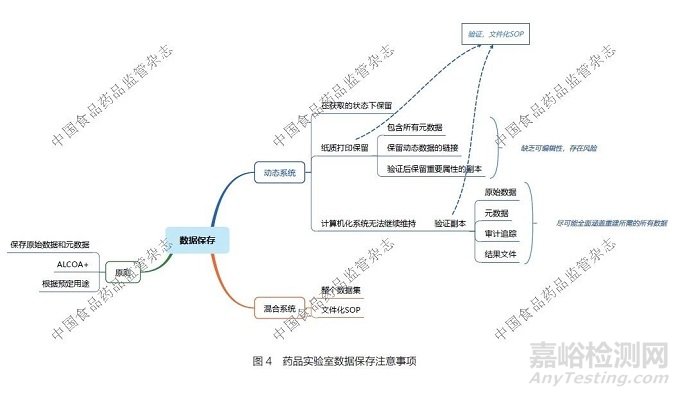
对于动态系统中的数据:①应在数据获取的原始状态下进行保留。②对于纸质打印保留的数据,要注意需包含所有元数据,且要保留动态数据的链接,或者经过验证后保留重要属性的副本,但纸质打印数据通常缺乏可编辑性,因此存在较高风险。③计算机化系统若无法继续维持运行,则应保存经过验证的副本。该副本应包含原始数据、元数据、审计追踪和结果文件等,尽可能全面涵盖重建所需的所有数据。④纸质打印数据和计算机化系统验证副本的过程都要经过验证,并形成文件化的操作规程。
对于混合系统的数据保存,需要对整个数据集(动态数据和静态数据)进行完整保存,并且保存的过程同样要经过验证,形成文件化的操作规程。
4.5 药品实验室数据管理软件主要验证考量要点
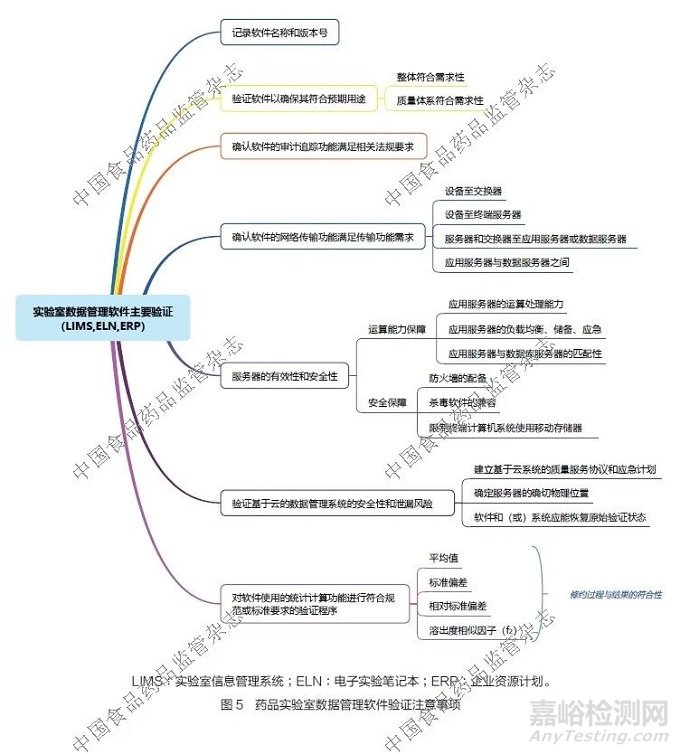
在实验室中,会使用到实验室信息管理系统、电子实验笔记本等数据管理软件。为确保数据完整性,需要对这些系统进行适用性验证,应重点关注以下要点:①记录软件名称和版本号。②验证软件以确保其符合实验室预期用途,包括整体符合需求性和质量体系具体符合需求性。③确认软件的审计追踪功能满足相关法规要求。④确认软件的网络传输功能满足实验室实际传输需求,包括设备至交换器、设备至终端服务器、服务器和交换器至应用或数据服务器、应用服务器与数据服务器之间等多个环节。⑤服务器的有效性和安全性验证。评估服务器的运算保障能力,包括应用服务器的运算处理能力、应用服务器的负载均衡、储备和应急能力,以及应用服务器与数据服务器的匹配性等, 确保其能满足实验室数据管理的计算需求;评估并确认服务器的安全保障,包括配备相应防火墙,考虑杀毒软件的兼容性,以及限制终端计算机系统使用移动存储器等。⑥基于云系统的验证考虑,包括验证数据管理系统的安全性和泄露风险,建立基于云系统的质量服务协议和应急计划,并确定服务器的确切物理位置,软件和(或)系统应能恢复原始验证状态等。⑦对系统软件使用的统计计算功能进行符合规范或标准要求的验证程序,不同的实验可以采用不同的验证参数,如平均值、标准偏差、相对标准偏差和溶出度相似因子(f2)等。在验证过程中,尤其要关注修约的次数和规则,及其对结果符合性判断的影响,特别是对临界数据的符合性判断。实验室数据管理软件验证注意事项详见图5。
05结 语
国内外药品监管机构、国际组织及行业协会对于药品实验室数据完整性的要求,既存在共性,又各具特色,并且在不同的GxP领域内还有相对细化的具体要求。随着对数据完整性认识的不断深入,笔者也对处于不同发展阶段的药品实验室在数据完整性方面提出了新的思考和完善相关体系的建议。具体到各实验室,需要根据自身的实际情况进行综合分析,切实理解并落实数据完整性的关键性原则做法,同时以文件化的形式,对重点环节的数据操作进行规范。其中,关键性原则做法就是要基于良好的数据完整性质量文化,在数据的全生命周期内,依托风险评估治理体系,确保数据具备ALCOA+ 全部属性。

来源:中国食品药品监管杂志