
嘉峪检测网 2024-10-23 17:18
导读:本文参考国内外药典、指导原则, 并结合美国食品药品监督管理局(FDA)各混悬型滴眼剂个例指导原则等, 对化学药品仿制药混悬型滴眼剂药学研究中应关注的内容进行了探讨。
摘要
混悬型滴眼剂系指难溶性固体药物以微粒状态分散于介质中形成的无菌眼用液体制剂。本文从化学药品仿制药混悬型滴眼剂的处方、生产工艺、质量研究、包材和稳定性研究等药学方面问题进行了初步的探讨, 汇总了国内外文献中混悬型滴眼剂开发过程中关键研究指标, 以助于业界对混悬型滴眼剂仿制药的开发。
关键词
混悬型滴眼剂; 仿制药; 药学研究
眼用制剂系指直接用于眼部发挥治疗作用的无菌制剂, 其中滴眼剂系指由原料药物与适宜辅料制成的供滴入眼内的无菌液体制剂, 可分为溶液、混悬液或乳状液。混悬型滴眼剂系指难溶性固体药物以微粒状态分散于分散介质中形成的无菌眼用液体制剂。
混悬型滴眼剂中活性成分以微粒状态分散于分散介质中, 相比溶液型滴眼液, 药物颗粒可在角膜前囊滞留, 增加了与角膜的接触时间, 有利于提高生物利用度[1]。目前国内外已上市多种混悬型滴眼剂, 如氯替泼诺混悬滴眼液、妥布霉素地塞米松混悬滴眼液、妥布霉素氯替泼诺滴眼液、布林佐胺溴莫尼定滴眼液、夫西地酸滴眼液、盐酸贝西沙星滴眼液等。
2023年2月, 国家药品监督管理局药品审评中心(CDE)发布了«化学药品仿制药溶液型滴眼剂药学研究技术指导原则»[2], 阐述了溶液型滴眼剂药学研究的要求。对于混悬型滴眼剂, 原料药不是以真溶液状态溶解于分散介质中, 相较于溶液型滴眼剂药学研究要求, 混悬型滴眼剂在处方、生产工艺、质量研究、包材和稳定性研究等方面有其特殊性。
本文参考国内外药典、指导原则, 并结合美国食品药品监督管理局(FDA)各混悬型滴眼剂个例指导原则等, 对化学药品仿制药混悬型滴眼剂药学研究中应关注的内容进行了探讨。
1、处方
对于溶液型滴眼剂, CDE发布的«化学药品仿制药溶液型滴眼剂药学研究技术指导原则»中, 建议仿制药处方中辅料种类(Q1)和用量(Q2)应与参比制剂一致, 其中辅料的用量相同是指仿制药辅料用量是参比制剂相应辅料用量的95%~105%。对于与参比制剂中渗透压调节剂用量、缓冲剂用量和pH调节剂不同的处方, 需标注不同之处, 阐述选择的理由, 并研究证明上述不同不影响产品的质量属性(如渗透压摩尔浓度、缓冲容量、pH值等物理化学特性)、安全性和有效性。
对于混悬型滴眼剂, 与溶液型滴眼剂一致, FDA各混悬型滴眼剂个例指导原则[3-9]中, 建议仿制药处方中辅料种类(Q1)和用量(Q2)一致, 辅料的用量相同是指仿制药辅料用量是参比制剂相应辅料用量的95%~105%。
通常, 已上市的国内外混悬型滴眼剂说明书中公开了辅料的种类。参考FDA公布的说明书, 表1中列举了部分混悬型滴眼剂中处方组成。
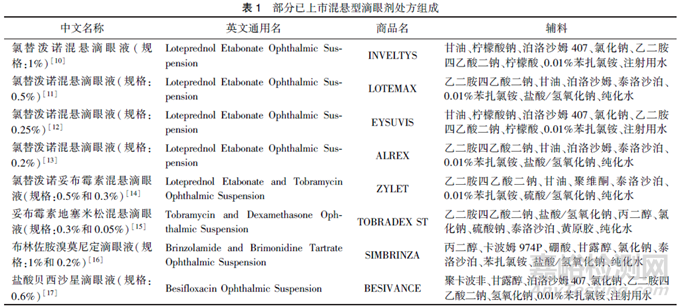
从FDA公布的已上市混悬型滴眼剂处方来看, 混悬型滴眼剂中辅料通常包括润湿剂、助悬剂、渗透压调节剂、pH调节剂、抑菌剂等。
在混悬型滴眼剂仿制药的处方开发过程中, 应基于参比制剂公开的说明书等信息, 确定原料药及辅料的种类, 同时应关注辅料的型号, 尤其是助悬剂等关键辅料的型号, 不同型号的辅料对混悬型滴眼剂质量可能有较大影响。如氯替泼诺混悬滴眼剂采用聚维酮作为助悬剂, 而市售聚维酮存在多种型号, 一般情况下应采用与参比制剂相同型号的辅料。
在对混悬型滴眼剂处方筛选过程中, 通常以渗透压摩尔浓度、粒度等作为考察指标, 通过合适的理化手段, 对辅料的用量进行筛选和解析, 以达到辅料的种类和用量与参比制剂一致。
多剂量滴眼剂一般应加适当的抑菌剂。对于处方中抑菌剂的用量, 在参考参比制剂用量基础上, 参照«中国药典»2020年版(四部)通则1121“抑菌效力检查法”, 对拟定处方的抑菌效力进行考察, 以确保抑菌效力符合药典抑菌效力检查法的规定, 并在产品整个生命周期符合质量要求。
2、生产工艺
滴眼剂为无菌制剂, 因此滴眼剂的生产也应采用无菌/灭菌工艺。不同于溶液型滴眼剂, 混悬型滴眼剂的原料药混悬于溶液中, 通常无法通过0.22μm的滤膜进行过滤除菌。目前, 混悬型滴眼剂通常有无菌生产和灭菌生产两种不同的生产方式:
①无菌生产: 由于原料药无法通过滤膜进行过滤除菌, 在生产中通常将辅料溶于注射用水, 过滤后制备得到辅料溶液, 然后将提前处理的无菌原料药加入辅料溶液中, 搅拌混合均质后, 无菌条件下灌装得到成品。
②灭菌生产: 灭菌生产通常包括两种不同的生产方式, 即浓溶液灭菌方式和稀配后灭菌方式, 两种方式的不同在于灭菌原辅料种类和灭菌体积的不同。对于浓溶液灭菌方式, 通常将原料药与部分辅料分散于部分注射用水中, 进行灭菌, 其他辅料和注射用水通过滤膜无菌过滤, 然后将灭菌后含有原料药的浓溶液与无菌过滤后的部分辅料溶液混合, 均质后, 无菌条件下灌装得到成品。对于稀配后灭菌方式, 通常根据原辅料性质, 按照一定顺序, 配制得到最终稀溶液, 灭菌后灌装得到成品。
基于以上混悬型滴眼剂的生产工艺, 在产品的工艺开发阶段, 应重点对原料药的前处理、原辅料加入顺序及方式、无菌过滤参数或灭菌工艺参数等进行考察和研究。
对于原料药的前处理, 原料药的粒度是混悬型滴眼剂的关键质量属性, 应对原料药的粒度分布进行考察和控制, 可采用激光衍射仪等对原料药的d(0.1)、d(0.5)和d(0.9)等进行测定和相应的控制。如采用微粉化方式对原料药进行前处理, 应关注微粉化后原料药的晶型变化。对于无菌原料药, 可选择采用无菌生产的原料药或采用辐照等方式对原料药进行灭菌处理。
对于混悬型滴眼剂的工艺验证, 除连续3批产品的工艺验证外, 一般还包括除菌过滤验证, 可参考国家药品监督管理局发布的«除菌过滤技术及应用指南»[18-19]进行研究和验证。对于生产中所用的除菌滤芯, 建议单次使用。另外, 在生产工艺开发中, 通常应对药液与生产组件的相容性进行研究[20], 以确认生产组件适用于产品的生产。
3、原料药和辅料
原料药应结合拟定的制剂生产工艺对其质量进行控制, 如粒度分布、晶型、无菌、微生物限度等。
对于辅料, 除进行常规质量控制外, 通常应对微生物限度进行控制。
4、质量研究
通常滴眼剂的质量属性包括性状、鉴别、溶液澄清度与颜色、渗透压摩尔浓度、pH值、黏度(如适用)、相对密度、可见异物、有关物质、元素杂质、无菌、抑菌剂含量、抗氧剂含量、含量测定、装量/装量差异等。对于混悬型滴眼剂, 还应关注原料药粒度和粒度分布、体外释放、滴出量、表面张力、沉降体积比、再混悬/再分散性、可溶性的药物量、含量均匀度等。部分研究项目讨论如下:
①原料药粒度和粒度分布: 原料药粒度和粒度分布是混悬型滴眼剂中关键质量属性, 混悬型滴眼剂质量研究通常应包含粒度和粒度分布的研究。氯替泼诺混悬滴眼液(规格: 0.5%和0.2%)等FDA个例指导原则中建议采用3批自制样品与3批参比制剂进行粒度与粒度分布对比, 样品的制备方法和粒度测定方法应进行验证以确保方法的准确性和可靠性, 同时在方法开发过程中应考察在不同稀释条件下对原料药粒度和粒度分布测定结果的影响, 测定参数为d(0.5)和SPAN[d(0.9)-d(0.1)/d(0.5)], 采用群体生物等效性(PBE)研究的分析方法进行d(0.5)和SPAN数据对比, 基于95%置信区间上限判定自制品与参比制剂的生物等效性。
②体外释放: 混悬型滴眼剂中原料药的体外释放可反映处方和工艺变化, 并可能影响药物的吸收程度。一般情况下, 体外释放方法应尽可能模拟实际临床的使用场景, 并兼顾平台期, 体外释放方法开发应包括释放介质种类和体积、取样时间点、温度的选择等。氯替泼诺混悬滴眼液(规格: 0.5%和0.2%)等FDA个例指导原则中建议采用3批自制样品与3批参比制剂进行体外释放(IVRT)对比, 同时采用恰当的统计方法(如非模型依赖相似因子比较法, f2)对体外释放数据进行比较分析。
③滴出量: 人的眼睛对于滴眼液的容量有限, 过高的滴出量会造成药物的浪费, 过低的滴出量可能影响药物的疗效。FDA«局部用滴眼剂的质量考虑»草案[21]中推荐仿制药每滴的滴出量应在参比制剂滴出量的±10%范围内, 且每滴的滴出量应为20~70μL范围内。
④可溶性的药物量: 通常混悬型滴眼剂中可能会有微量或少量的原料药溶于溶液中, 可溶性的药物量除可比较自制品与参比制剂质量外, 还可反映药物处方、工艺设计的合理性。氯替泼诺混悬滴眼液(1%规格和0.25%规格)等FDA个例指导原则中建议对可溶性药物量进行对比研究。
⑤再混悬/再分散性、含量均匀度: 再混悬/再分散性是混悬型滴眼剂重要质量指标, 在质量研究中, 应结合参比制剂说明书使用方法, 如使用前是否需摇匀等, 对再混悬/再分散性等特性进行对比研究。同时FDA«局部用滴眼剂的质量考虑»草案中建议对多剂量混悬型滴眼剂含量均匀度进行测定, 通常应对滴眼剂瓶上部、中部和底部药物的含量进行测定, 以确证原料药是否均一地分散且在整个有效期内可被均一地递送。
5、包材研究和稳定性研究
混悬型滴眼剂仿制药, 应根据参比制剂所用包材和产品特点选择合适的包装材料。原则上, 所选择的内包材在对产品保护性、功能性方面, 应不低于参比制剂所用包材。同时应考察产品与包材的相容性, 并对包材的密封性进行研究。包材的密封性可参考«化学药品注射剂包装系统密封性研究技术指南(试行)»[20]开展研究。
对于稳定性, 应结合包材的特点(如半透性包材等)设计相关的稳定性试验, 在稳定性考察期间, 除一般质量研究项目外, 还应对原料药粒度与粒度分布、沉降体积比、再混悬/再分散性、含量均匀度、抑菌剂含量、抗氧剂含量、失水率等进行考察, 必要时应考虑对原料药体外释放进行考察。稳定性试验样品通常应包括直立放置和倒置(或平放)两种放置方式。
对于混悬型滴眼剂, FDA«局部用滴眼剂的质量考虑»草案中要求, 应进行冷冻/解冻热循环研究, 以评估运输和处理过程中可能遇到的任何高温和低温变化的影响, 这些变化可能影响药品的质量。FDA建议该研究由3个循环组成, 温度在冷冻(-20℃至0℃)和环境温度(25℃至35℃)之间循环, 累计最少3d。在整个研究过程中, 以及在预定的周期数结束时, 应对样品所有质量研究项目进行分析, 并与参比制剂进行比较。
对于多剂量滴眼剂, 还应考察其使用中的稳定性。
6、小结
混悬型滴眼剂作为较复杂的滴眼剂型, 其仿制药的开发除遵循一般滴眼剂的开发路径外, 还需关注其特殊性, 如粒度和粒度分布、含量均匀性等。
目前, 国内外监管机构暂未发布专门的关于混悬型滴眼剂研究的指导原则, 仿制药的开发存在一定的难度, 国内上市的混悬型滴眼剂仿制药较少, 本文对混悬型滴眼剂仿制药开发中相关的药学问题进行了探讨, 以供业界参考。

来源:凡默谷