
嘉峪检测网 2025-03-06 09:17
导读:本文在梳理已上市化学药品注射剂生产批量变更相关指导原则及技术要求的基础上,从关联变更、工艺验证、质量对比研究等方面阐述变更的研究思路.
摘要:本文在梳理已上市化学药品注射剂生产批量变更相关指导原则及技术要求的基础上,从关联变更、工艺验证、质量对比研究等方面阐述变更的研究思路,结合药品上市后注册管理事项中等变更日常审查工作实践,总结变更管理中的变更信息描述不全、验证不符合相关指导原则要求、质量对比不全面、稳定性研究不规范等常见问题。
注射剂是一种由原料药与适宜辅料制成的供注入人体内的无菌制剂[1],具有直接进入血管、组织或器官,具有吸收快、作用迅速的特点。尤其是静脉注射剂中的药物能直接进入血液循环发挥药效,故而在药品监管和临床使用中被视为高风险剂型。在实际生产过程中,因扩线增产、生产设备技术升级、市场供应等多种因素,化学药品注射剂生产批量的变更较为常见。但无论何种原因导致的批量变更,都必须确保制剂质量不受影响,这是药品变更管理的关键所在。
为有效规范药品上市后变更管理工作, 保障公众用药安全, 国家药品监督管理局及其审评中心在2021 年相继出台多项政策法规。1月12日发布的《药品上市后变更管理办法(试行)》,依据《药品管理法》落实了药品生产变更的分类管理要求,明确变更原则、情形与各方职责,为变更管理奠定基础;2 月10日颁布的《化学药品变更受理审查指南(试行)》和《已上市化学药品药学变更研究技术指导原则(试行)》(以下简称《指导原则》),则进一步明确化学药品批量、工艺等变更细节及研究验证工作,为企业和监管部门在实际操作中提供了关键的指导规范。
自这些政策实施以来,企业在依据政策开展化学药品注射剂生产批量变更的实际操作中,仍存在一些问题。鉴于此,本文通过梳理已上市化学药品注射剂生产批量变更的相关指导原则与技术要求,并结合日常中等变更审查工作实际,剖析注射剂变更研究的思路与常见问题,旨在为企业变更和监管部门审查提供参考, 保障药品变更过程科学规范,进而维护公众用药的安全有效。
1. 技术要求
根据《指导原则》,已上市化学药品注射剂在进行变更时,应遵循以下原则:①药品上市许可持有人作为变更研究的主体,应承担起设计和开展变更研究工作的重任, 全面评估变更对药品的影响;②应关注批量变更引发的关联变更所导致的叠加影响;③变更研究应采用基于商业化规模生产的样品展开,以确保各项研究(如工艺研究与验证、相容性研究、密封性研究等)严格遵循相关要求,并能真实反映实际生产情况。
1.1 变更情形
《指导原则》对注射剂批量变更在注册管理类别要求上有明确要求[2]:同时,该《指导原则》提及“变更不影响制剂关键质量属性的工艺参数” 属于中等变更;然而在药品上市许可持有人的药品变更管理实践中,对于工艺参数变更的注册管理类别判定仍存在不少困惑。实际上,不同工艺参数变更,因风险差异,其注册管理类别也不尽相同。例如国际药品管制协调会(ICH)制定的Q12 标准中“确定生产工艺参数既定条件和相关报告类别的决策树”为工艺参数变更管理提供了一种思路,可从“是否需要对参数进行控制”“对产品质量产生潜在影响”等方面确定具体某项工艺参数变更的类别[3]。
1.2 研究验证工作
化学药品注射剂批量变更所需开展的研究工作包括:①说明批量变更详情及原因, 对比分析变更前后生产工艺及设备的设计与工作原理并对变更后的批量进行研究和/或验证。对于中等变更和重大变更,鉴于注射剂为无菌制剂,必要时需进行无菌/灭菌工艺验证。②提供变更后一批样品的批生产记录。③对变更前后样品开展质量对比研究,确保杂质谱、关键理化性质等保持一致且符合相关要求。④对变更后三批样品进行检验,确保符合质量标准规定。⑤微小变更对变更后首批样品进行长期稳定性考察,并在年报中报告其长期稳定性试验数据;中等变更要对变更后一批样品进行加速及长期稳定性考察,申请时提供不少于3 个月的稳定性研究资料并与变更前产品比较,确保变更后样品的稳定性不低于变更前;重大变更对变更后三批样品进行加速及长期稳定性考察, 申请时提供3 ~ 6 个月的稳定性研究资料并与变更前比较,保证变更后产品的稳定性不低于变更前。⑥重大变更还需依据变更情况综合评估是否开展生物等效性研究。如申请免除,需结合工艺复杂度、药物特性、批量变更情况及生产设备等因素综合考虑,提供充分依据。
1.3 特殊要求
特殊注射剂批量变更需予以特别关注。相较于普通注射剂,特殊注射剂的质量及活性成分的体内行为受处方和工艺影响显著,可能影响其体内的安全性和有效性,如脂质体、静脉乳、微球、混悬型注射剂、油溶液和胶束等。鉴于制剂特性的复杂性,为证实特殊注射剂仿制药与参比制剂的质量和疗效一致,注册研发时除药学研究外,还需要开展非临床、人体生物等效性研究,乃至临床研究。由于复杂工艺变更和批量放大可能影响特殊注射剂的质量和疗效,根据《指导原则》,特殊剂型制剂生产批量变更属于重大变更,因此对于复杂工艺生产的特殊注射剂,注册批生产规模应与拟定商业化生产规模一致[4]。对于存在明确安全性问题的品种,如多组分生化药注射剂、中西药复方注射剂等[5],不宜再进行相关的变更研究,同时需关注原批准品种继续生产的安全性风险。
2. 研究思路
2.1 批量变更总体思路
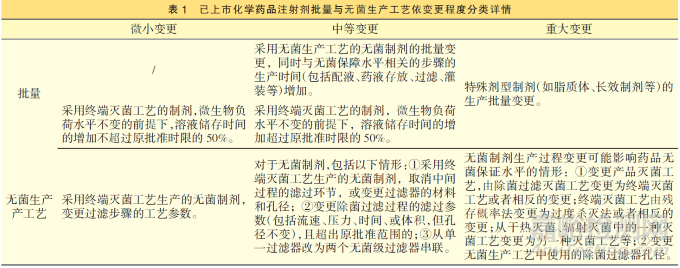
《指导原则》明确了化学药品注射剂批量变更条件,见表1,如无菌生产工艺步骤的生产时间是否增加; 终端灭菌工艺的制剂在微生物负荷水平不变的前提下, 溶液储存时间的增加是否超过原批准时限的50%等。值得注意的是,为避免多次变更的叠加影响, 上述生产时间的计算均为注册批准时关键临床批、工艺验证批次等代表性批次进行对比,以确定变更管理类别。确定类别后,药品上市许可持有人应按相应研究验证工作要求开展工艺验证、无菌/灭菌工艺验证、质量与稳定性对比研究等,以证明变更未影响产品安全性、有效性和质量可控性。
2.2 综合评估关联生产设备及工艺参数变更的影响
批量变更往往伴随着生产设备或工艺参数改变,如某小容量注射剂批量增加,关联生产设备变更包括配液罐型号变更,并关联生产工艺参数变更包括搅拌转速及搅拌时间、过滤时间、灌装时间增加等。因此,《指导原则》要求“对变更前后的生产工艺及生产设备的设计及工作原理进行对比分析”。
研究重点关注:①关联设备变更:全面比较变更前后生产设备生产厂家、型号、材质、设备原理、关键技术参数, 确认与生产工艺的匹配性。如灭菌柜变更, 变更前为脉动真空灭菌柜, 变更后为水浴灭菌柜,两者灭菌原理和工艺参数不同,升温降温过程不尽相同,需开展全面灭菌工艺研究和验证工作;注射剂一致性评价要求应根据溶液的特点和生产工艺对直接接触药液容器的相容性进行研究,某注射剂批量变更关联了生产管路材质的变更,由不锈钢变更为硅胶管,应重新对药液与硅胶管的相容性进行研究。②关联工艺参数变更:基于“质量源于设计”的理念,针对可能影响药品关键质量属性的关键工艺参数进行充分研究。例如混合工序的规模放大是常见且需要重点关注的问题,可参考《制剂工艺放大》采用流量型搅拌强度法,此方法规定大小规模设备具有相同的液体速率, 可以通过变更前后混合罐搅拌叶轮制直径、搅拌槽直径、雷诺数等计算得到变更后混合罐所需的搅拌转速[6]。③工艺参数优化:如某注射剂活性成分易氧化降解, 注册申报时采用过量投料, 批量放大同时将生产设备由半密闭系统变更为全密封系统,且残氧量控制限度收紧,使得变更后样品更加稳定,可以取消过量投料。
2.3 对变更后的批量/设备/工艺进行充分验证
批量变更涉及各个工序,需进行全面的生产工艺验证和灭菌/无菌工艺验证,不能简单照抄首次注册时的验证方案。一方面应参照国内外最新相关指导原则,如国家局于2018 年发布了《除菌过滤技术及应用指南》《无菌工艺模拟试验指南(无菌制剂)》等指导性文件,进行差距分析,依变更后的生产批量、设备、参数制定适配的验证方法。另一方面,应关注批量变更后的生产情况是否超出了原验证范围,例如:①无菌/灭菌验证的样品代表性。如《无菌工艺模拟试验指南(无菌制剂)》中要求“对于超大批量的生产规模,例如大于100 000 支,则应考虑适当增加模拟的灌装数量”,如某小容量注射剂,生产批量由8 万支增加到50 万支,培养基模拟灌装数量仍为10 000 支,未充分评估批量增加带来的无菌风险[7]。②某注射剂采用除菌过滤工艺,批量变更引起的过滤时间、过滤批量增加可能会影响细菌截留试验的结果, 因此需重新评估细菌截留试验工艺参数。③对于热敏感品种,如批量放大导致灭菌柜型号、容积变大,在热分布和热穿透等湿热灭菌工艺验证时还应关注冷热点的F0值,以及升温和降温速率的影响,也应对热点样品进行取样检验和质量对比研究[5]。
2.4 质量及稳定性对比研究
变更应开展质量及稳定性对比研究,考察项目依药物、注射剂特性以及批量、工艺变更的具体情况综合评估, 至少包括溶液颜色与澄清度、pH 值、有关物质(杂质谱变化情况、各单个杂质含量、总杂质含量)和含量等。冻干制剂还应考虑物理外观和复溶性。杂质谱对比研究中,应结合注册申报时强制降解试验的稳定情况,关注热不稳定、易氧化药品变更后新杂质引入,评估和验证方法适用性。在稳定性对比中, 各考察时间点应及时取样检测,汇总分析变更前后结果,如有异常趋势,应立即启动超趋势结果调查。
3. 常见问题
3.1 申报资料
药品上市许可持有人应参照《药品注册申报资料格式体例与整理规范》《指导原则》《化学药品变更受理审查指南(试行)》等要求提交备案资料,确保备案信息真实、准确、完整。
常见问题包括:变更情况描述过于简单,如:某产品批量变更,未按照指导原则提供工艺及生产设备的对比分析资料;部分产品虽提及无菌保障水平相关步骤的生产时间增加,但未提供变更前后从配液开始到除菌过滤开始、除菌过滤开始至灌装结束的具体时间,无法判断无菌模拟灌装验证时常是否覆盖相关工序;缺少工艺验证方案或报告;缺少变更后三批样品的检验报告等。
3.2 工艺验证、无菌/灭菌工艺验证
应按照最新技术指导原则开展验证工作,通过风险评估确定验证的范围,合理设计额外试验和可接受标准,并分析总结验证结果。
常见问题包括:无菌分装产品变更后,缺少培养基模拟灌装试验资料;工艺验证参数超出范围却未说明理由;工艺验证报告缺少关键工艺参数数据和中间体检测数据。
3.3 质量对比研究
需提供质量对比研究的样品信息、检测项目、检测结果及对比结果分析。
常见问题包括:样品信息不全,如无生产时间、生产线、批量等;采用中试规模代替商业化生产规模样品进行质量对比且无充分依据;未提供变更前样品生产信息,难以确定其与一致性评价获批时是否一致。检验报告书信息有误:如检验项目和注册标准或内控标准中检验项目不一致;多批次检验依据不同且未说明质量标准变更情况。
3.4 稳定性研究
应按照指导原则要求开展稳定性试验,提供稳定性研究方案、变更前后结果及后续考察承诺。
常见问题包括:缺少变更前后稳定考察条件;或条件不符合《中国药典》及相关指导原则中规定温、湿度;变更前后条件不同,直接对比数据,无合理说明。考察项目不全面,如:为考察质量标准中溶液澄清度和颜色、细菌内毒素、不溶性微粒、晶型等,但长期稳定性等指标考察频次不合理,如对注射剂稳定性试验重点考察项目不溶性微粒, 加速、长期稳定性试验方案要求的检查时间点为加速第0、6 个月,长期第0、24、36 个月。该考察频次不能充分反映产品稳定性状况;变更前后的稳定性考察结果存在异常情况,如杂质检测结果波动大,高于变更前,或变化趋势不同等。
3.5 注册管理类别
特殊注射剂批量变更可能影响制剂在体内的安全性和有效性。根据《指导原则》属重大变更,若按中等变更备案则依据不足。
4. 展望
无菌制剂中不能含有活微生物且应满足内毒素限度标准;由于无菌与内毒素问题的可检测性低,一旦出现问题易对患者造成危害, 故无菌过程的风险管理至关重要[8]。如欧盟2022 年修订的无菌附录中提及的污染控制策略(CCS),批量及关联工艺变更若影响CCS 污染控制风险水平,需进行风险审查与评估[9]。建议参考ICH Q9《质量风险管理》工具评估无菌保障风险,PDA 的TR44《无菌过程质量风险管理》介绍了无菌制剂生产中的风险管理工具与实例,如用FMEA 评估冻干瓶制剂轧盖等风险,用风险评分数评估无菌灌装工序风险。
药品上市许可持有人应履行好药品上市后变更管理的主体责任,不断加深对产品关键生产工艺、关键质量属性、无菌保障等认识理解,并在此基础上开展相应的研究及验证工作, 根据评估的结果进行补充申请、备案或年报,以确保变更对药品的安全性、有效性和质量可控性没有产生不良影响。同时鼓励药品上市许可持有人采用ICH 指导原则(如ICHQ12 等)中的各种变更管理工具,如既定条件、批准后变更管理方案等,对变更进行研究和分类,这将更有利于主动对已上市药品进行持续改进和创新。省级药品监管的技术支撑单位将进一步加强药品上市后变更相关问题的政策解读、案例和常见问题分享,畅通沟通交流渠道, 逐步发布中等变更的申报资料要求和审查标准,以便指导药品上市许可持有人更加科学、规范开展相应的研究和验证工作。
参考文献
[1] 国家药典委员会. 中华人民共和国药典: 三部[S]. 北京:中国医药科技出版社, 2020: 441.
[2] 国家药品监督管理局药品审评中心. 已上市化学药品药学变更研究技术指导原则(试行)[S]. 2021.
[3] ICH Q12: Technical and regulatory considerations forpharmaceutical product lifecycle management.https://database.ich.org/sites/default/files/Q12_Guideline_Step4_2019_1119. pdf
[4] 章俊麟, 石勇平, 那馨竹, 等. 化学药品特殊注射剂仿制药药学研究技术要求[J]. 中国新药杂志, 2023, 32(2): 163-7.
[5] 蒋煜, 马磊. 注射剂变更申请的研究思路及案例分析[J]. 中国医药工业杂质, 2016, 47(3): 355-62.
[6] [美]M. 莱文(M.Levin)主编, 唐星译, 制剂工艺放大[M].北京: 化学工业出版社, 2020: 35-50.
[7] 国家药品监督管理局. 无菌工艺模拟试验指南(无菌制剂)[S]. 2018.
[8] PDA Technical Report No.44: Quality Risk Managementfor Aseptic Processes. https://www.pda.org/bookstore/ product-detail/1217-tr-44-quality-risk-management-foraseptic
[9] PDA Technical Report No.90: Contamination Control Strategy Development. https://www.pda.org/bookstore/product- detail/7155-technical-report-90-contamination-control
内容来源:药学与临床研究

来源:Internet
关键词: 注射剂