
嘉峪检测网 2025-04-21 20:42
导读:本文介绍了台湾医疗器械注册认证指南。
一、基本概况
1.自然环境
台湾地区的面积为3.6万平方公里,是中国不可分割的一部分。台湾西部与西北部临台湾海峡,与福建省隔海相望,距大陆海岸平均距离约200公里,台湾海峡最窄处为新竹县到福建省平潭岛,直线距离约130公里;北边隔东海与朝鲜半岛相望;东北隔海与琉球群岛相望;西南为南海,距广东省海岸约300公里;东边为太平洋,和日本冲绳县与那国岛相邻不到110公里;南边则隔巴士海峡与菲律宾群岛相邻。
2.人口和行政区划
截至2024年12月底,台湾全省常住人口2340.02万人。人口数量超过200万以上的城市有新北市(404.7万)、台中市(286.06万万)、高雄市(273.14万)、台北市(249.08万)、桃园市(233.86万)。台湾人普遍使用汉语普通话和繁体中文,另外比较常见的语言有闽南语、客家语和台湾高山族语言。常见使用的外语为英语和日语。
台湾目前包括6个“行政院直辖市”、3个市、13个县。共下辖146个乡、38个镇、14个县辖市、170个区。
3.2024年出口概况
2024年,中国向中国台湾出口医疗器械总计约34.14亿元人民币,同比上升11.29%,增势可观。
二、监管机构和法规要求
中国台湾行政院卫生署食品药品管理局负责台湾的医疗器械产品注册;台湾医疗器械注册需要遵循如下法规要求:
◆ 《医疗器材管理法》
◆ 《医疗器材管理法施行细则》
◆ 《医疗器材分类分级管理办法》
◆ 《医疗器材品质管理系统检查及制造许可核发办法》
◆ 《医疗器材许可证核发与登录及年度申报准则》
三、医疗器械定义
根据《医疗器材管理法》第3条,医疗器械(醫療器材):指仪器、器械、用具、物质、软件、体外诊断试剂及其相关物品,其设计及使用是以药理、免疫、代谢或化学以外的方法作用于人体,而达到下列主要功能之一者:
①诊断、治疗、缓解或直接预防人类疾病;
②调节或改善人体结构或机能;
③调节生育(受孕控制)。
根据《体外诊断医疗器材查验登记须知》,体外诊断器械(體外診斷醫療器材In Vitro Diagnostic Device, IVD):指收集、处理或检查取自人体的样本,作为诊断疾病、判断健康状态或其他状况,而使用的诊断试剂、仪器、软件或系统;而体外诊断试剂是指前述的任何试剂、校准品或质控品。
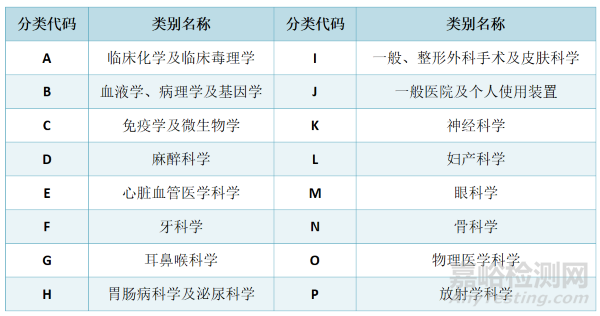
四、产品分类
依据《医疗器材分类分级管理办法》,医疗器械依功能、用途、使用方法及工作原理,视其应用科室分为16大类,并依其风险程度,分为以下三个类别:Ⅰ类/第一等级(低风险)、Ⅱ类/第二等级(中风险)及Ⅲ类/第三等级(高风险)。
五、注册流程
1.注册流程图
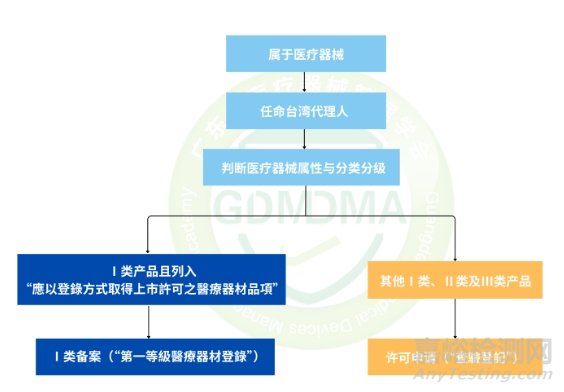
流程图解释:
◆ QSD符合性申请:准备质量体系文件(QSD)→递交QSD审查申请及缴费→TFDA审查及补正通知→资料补正(2个月内)→TFDA批准签发QSD证书;如涉及极高风险产品需启动现场审核,请参考“5、现场审核要求及注意事项”。
◆ 产品注册申请:准备注册申请文档→递交注册申请及缴纳审查费用→TFDA受理→形式审查→发补补正(4个月内)→实质性审查→发补补正(3个月内)→TFDA批准及发送审查结果通知书→申领证书及缴纳证书费→TFDA签发证书;
◆ QSD证书有效期3年,到期前6个月至12个月内提出续证申請;医疗器械许可证有效期为5年,到期前6个月内提出续证申请。
2.注册提交文档
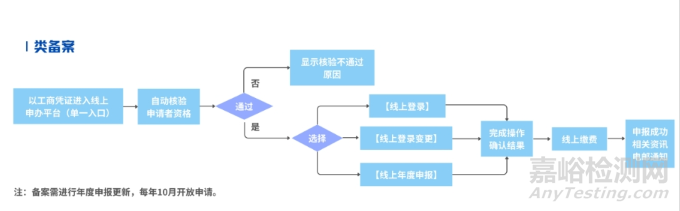
Ⅰ类备案
填报资料包括:台湾代理人信息、制造商信息及产品基本信息,并上传资料(如ISO 13485证书、医疗器械制造商制造业(生产)许可证复印件等)。
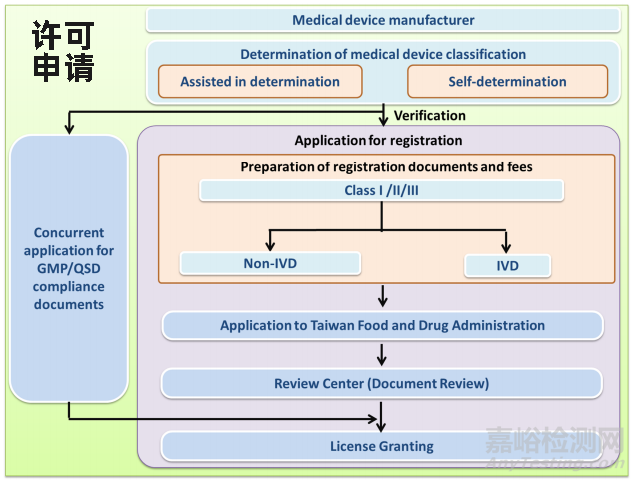
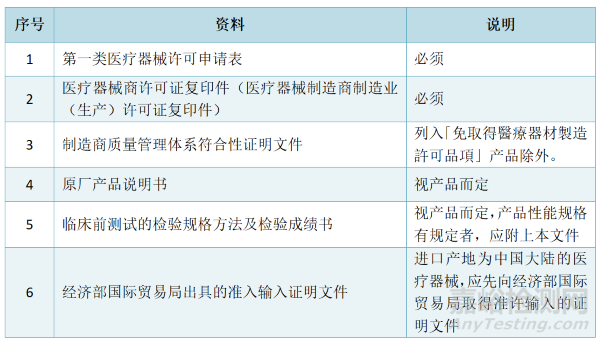
Ⅰ类许可
Ⅱ类及Ⅲ类许可
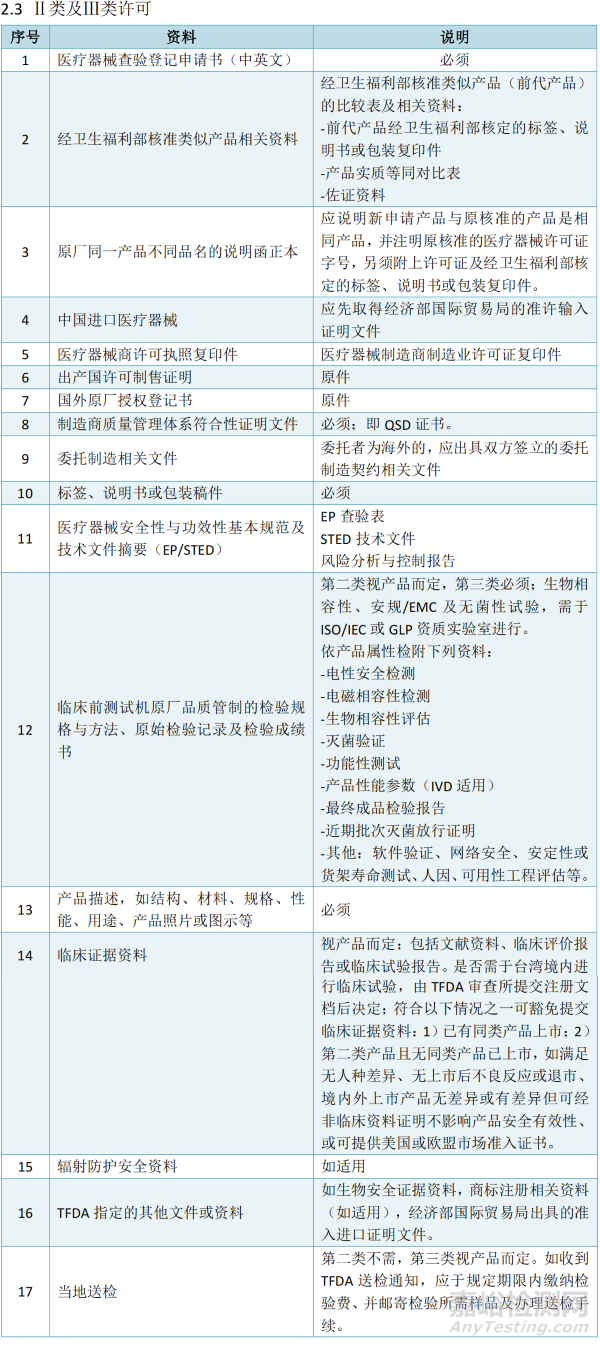
注:IVD产品另需额外考虑
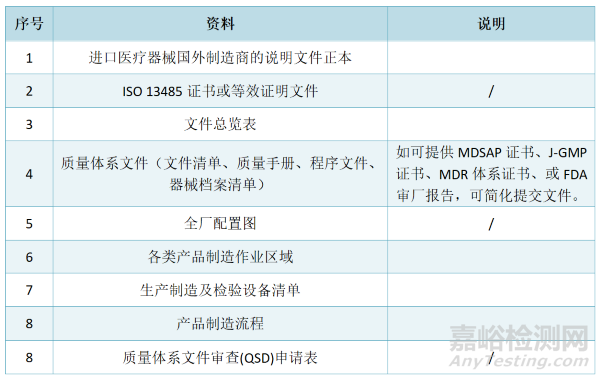
质量体系文件审查(QSD)申请
3.注册周期及费用
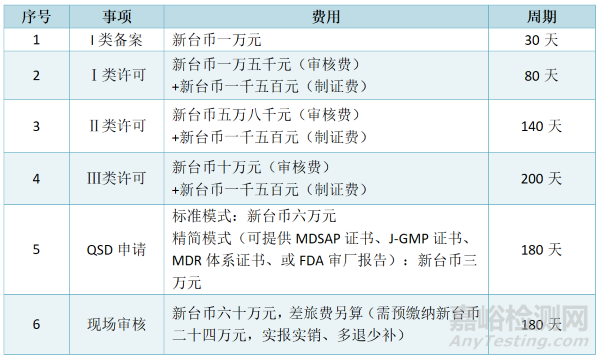
注:如涉及临床试验资料审核,费用另计;其中临床试验方案-新台币五万元,临床试验报告-新台币五万元,临床试验文件技术性评估-新台币两万元,临床试验变更审查-新台币五千元,海外临床试验机构现场核查-新台币六十五万元。
4.注册申办平台
Ⅰ类:第一等級醫療器材線上登錄
Ⅱ类、Ⅲ类:第二、三等級醫療器材查驗登記電子化送件
QSD及现场审核申请:醫療器材品質管理申請平台
5.现场审核要求及注意事项
极高风险产品
如属于下列极高风险产品,则需启动现场审核。
1. 心脏瓣膜置换物(E.3925)
2. 血管移植弥补物(E.3450)
3. 角膜弥补物(M.3400)
4. 被动式肌腱弥补物(N.3025)
5. 植入式心律器之脉搏产生器(E.3610)
现场审核流程
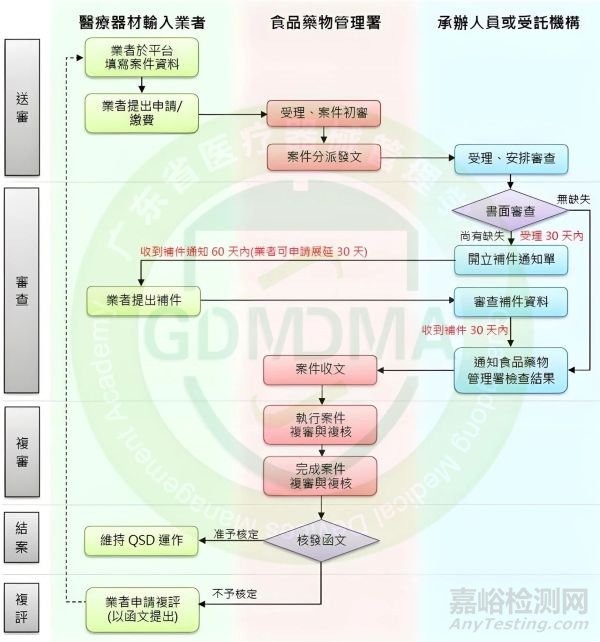
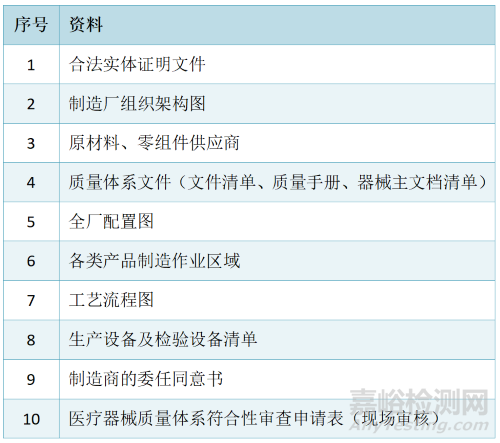
现场审核申请资料清单
6.该区域有关UDI的要求
TFDA已针对第三类植入式(2021年6月)、第三类非植入式(2022年6月)及第二类(2023年6月)强制实施了UDI规定。
赋码
除了下表中所列举第二类医疗器械可豁免赋码PI,其他器械需赋码至PI信息;
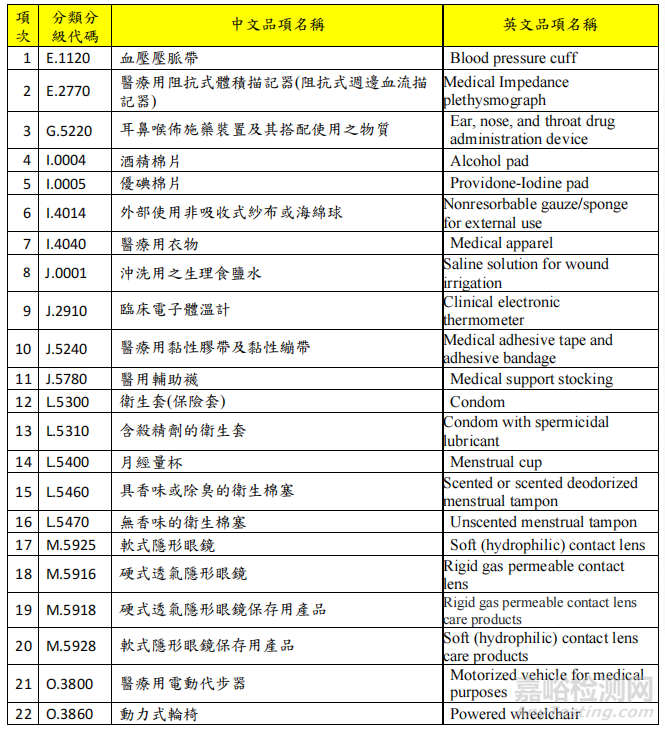
上传
台湾代理人应于产品上市流通前,将UDI - DI及相关资料上传。
溯源
此外,针对某些特定植入类高风险产品,TFDA还规定了「醫療器材來源流向資料」申报要求;即台湾代理人应于每年一月、四月、七月及十月之二十日前以电子方式申报产品来源及流向或销售使用信息,申报平台为“醫療器材來源流向資料申報平台”,需申报产品种类如下:
1. E.3610植入式心律器之脉搏产生器。
2. I.3540硅胶充填之乳房弥补物。
3. L.5980经阴道骨盆腔器官脱垂治疗用手术网片。
7.台湾代理人
资格条件、角色和职责
◆ 资格条件
台湾代理人必须为在台湾成立的法人实体,并持有营业执照和医疗器械经营许可证(“醫療器材商許可執照”)。
◆ 角色与职责
1. 台湾代理人(“醫療器材許可證所有人或登錄者”)为许可证所有人,代表制造商向台湾TFDA递交所有注册文件;
2. 协助获取质量体系文件(QSD)证书(此要求针对II类和III类以及部分I类医疗器械);
3. 作为制造商和TFDA之间的法规事物联络人;
4. 监管医疗器械的严重不良事件(SAE)报告;
5. 协助维护QSD证书和医疗器械备案或许可证书。

来源:广东省医疗器械管理学会
关键词: 医疗器械注册