
嘉峪检测网 2025-04-25 20:28
导读:本文介绍了Crinecerfont胶囊CMC变更桥接评估。
|
药品名 |
CRENESSITY (Crinecerfont) |
|
剂型 |
胶囊 |
|
规格 |
25 mg, 50 mg, 100 mg |
|
使用途径 |
口服 |
|
Rx/OTC |
Rx处方药 |
|
适应症 |
适应于糖皮质激素替代的辅助治疗,以控制4岁及以上患有经典先天性肾上腺增生的成人和儿科患者的雄激素 |
|
最大日服用剂量 |
200mg |
Crinecerfont属于新化学分子,其特性鉴定、纯度、杂质及生产工艺均通过适当质量管理体系和验证的分析方法得到有效控制。原料药在商业包装内于规定贮存期内稳定性良好。
该新药为口服软胶囊剂型,辅料成分包括:中链甘油三酯、二辛酸/二癸酸丙二醇酯、月桂酰聚氧-32甘油酯及维生素E聚乙二醇琥珀酸酯。
需说明的是二辛酸丙二醇酯虽首次应用于口服制剂,但根据药学/毒理学审评意见确认其在处方中的定量水平可接受。药品规格分为25mg、50mg和100mg三种,采用儿童防护盖的HDPE瓶包装。生产过程由cGMP合规生产设施的质量管理体系有效管控。
从质量角度评估,拟定的控制策略能够充分保证产品在特性、规格、纯度、效价及稳定性方面的质量一致性。
稳定性期间的IVR数据:
使用拟定的溶出度方法/接受标准对三个规格的代表/临床批次进行了稳定性研究,如表8A–8C所示。主要稳定性数据是在长期(25°C/60% RH)、加速(30°C/65% RH)和更严苛的(40°C/75% RH)存储条件下生成的。
FDA评估:
如上表7所示的溶出度方法和验收标准已建议在放行时及整个稳定性期间对药品进行质量控制。

B.3. IVR方法的临床相关性及接受标准(如IVIVR/IVIVC、计算机建模、小规模体内研究):
FDA评估:
申请人未进行IVIVR/IVIVC或任何模型研究。尽管拟定药物为速释制剂,其达峰时间范围从空腹状态7.0小时至餐后5.0小时。速释制剂较长的达峰时间可能归因于crinecerfont原料药属于低溶解度/低渗透性(BCS 4类)物质。基于申报数据,拟定的溶出度方法及接受标准未体现临床相关性,主要服务于保障批间一致性,此标准可接受。
制剂的稳定性考察了3种规格各3批,稳定性条件是ICH常规的要求条件。虽生物等效性研究没有在25mg和50mg规格上进行,但稳定性研究不能省略,即使三种规格是完全等比例处方,但化学稳定性还是需要考察,没有使用括号法节省的方式。
B.12. 制剂的桥接:
审评员的评估:
软明胶胶囊是最终的(TBM)商业经制剂。拟上市的TBM药品分为三种规格(25 mg、50 mg和100 mg)。下表9A显示了TBM制剂三种规格的组成。三种规格的胶囊填充制剂在组成成分上相互成比例。
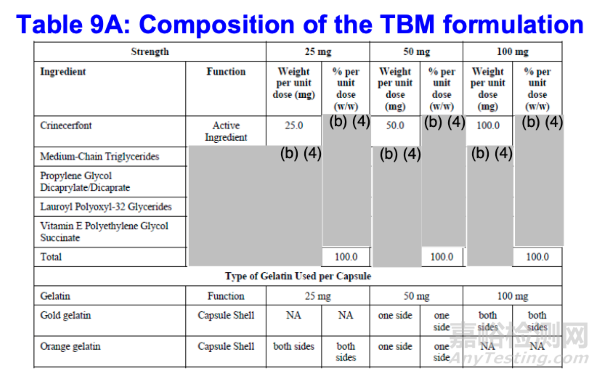
III期研究使用的是50毫克硬明胶胶囊。胶囊填充配方的定性和定量组成在III期胶囊配方和TBM配方中完全相同(表9B)。
一项I期比较生物利用度研究(研究编号NBI-74788-1736)旨在桥接TBM制剂100 mg规格与III期50 mg硬明胶胶囊。药代动力学参数(Cmax、AUC0-tlast和AUC0-inf)几何平均值的90%置信区间符合0.8-1.25的接受标准范围(表10)。研究编号NBI-74788-1736的评估由OCP/临床药理学审评员负责。尽管2×50 mg III期胶囊与1×100 mg TBM胶囊的溶出数据比较显示f2<50(表10),FDA认为,由于比较溶出研究采用的是拟定溶出方法(表7),而该溶出方法是为TBM软明胶胶囊优化的方案,可能不适用于III期硬明胶胶囊。

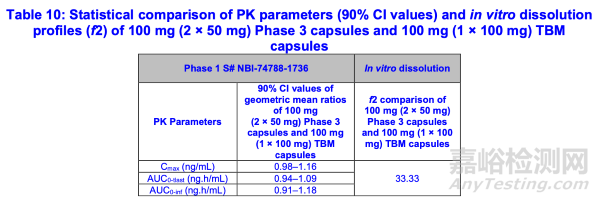
根据PK参数(Cmax、AUC0-tlast和AUC0-inf)的90%置信区间值符合0.8-1.25的接受标准范围,TBM产品100 mg规格(软明胶胶囊)可认为已成功桥接至III期临床用50 mg规格的硬明胶胶囊。
对于其他/较低规格(25 mg和50 mg)的桥接研究:
已对100 mg规格的TBM制剂进行了1期PK研究(研究编号NBI-74788-1736)。根据21 CFR 320.22(d)规定,申请人提出基于以下条件在25 mg和50 mg规格的TBM制剂与100 mg规格的TBM制剂之间建立桥接:
1)单次口服crinecerfont胶囊制剂(25至200 mg)后,其暴露量参数(Cmax和AUC)在临床相关剂量范围内具有剂量比例性;
2)BM胶囊100 mg、50 mg和25 mg规格的制剂生产工艺以及定性与定量组成具有比例关系;
3)注册用25 mg和50 mg胶囊分别与100 mg TBM胶囊及2×50 mg三期临床胶囊具有相似的体外溶出曲线;
4)比较使用的参比制剂(100 mg TBM胶囊)已在体内临床研究中证实与三期临床胶囊具有生物等效性。
对比确证性临床所用50mg规格的硬胶囊时,100软胶囊的溶出数据与50mg硬胶囊不相似,f2<50. 原因是胶囊壳理化性质不完全一样,产生了不一样的溶出数据。根据体内的PK数据对比,两种性质的胶囊制剂可以达到0.8-1.25,因此根据体内的数据判断它们是可桥接的,不影响体内的性能表现。
其它的不同规格桥接包括了体内PK数据,工艺对比,处方组成比例对比,体外的溶出对比,体内生物等效性数据对比。
FDA评估:
针对100 mg规格的TBM制剂进行了I期研究。根据21 CFR 320.22(d)(2),为支持25 mg和50 mg规格TBM制剂与100 mg规格TBM制剂之间的桥接,申请人提交了以下数据证明:
1)口服crinecerfont后暴露量参数(Cmax和AUC)在胶囊制剂(25至200 mg)的临床相关剂量范围内呈剂量比例关系
2)100 mg、50 mg和25 mg规格TBM胶囊的灌装制剂生产工艺、定性定量组成均具比例关系(表9A)
3)体外比对研究中使用的参比制剂(100 mg TBM胶囊)已通过体内临床研究(研究编号NBI-74788-1736;表10)证实与III期胶囊具有生物等效性
4)表11显示了TBM 100 mg胶囊(1×100 mg)与注册规格TBM 25 mg(4×25 mg和1×25 mg[标示量%])及50 mg(2×50 mg和1×50 mg[标示量%])胶囊的体外溶出曲线数据比较(f2值)
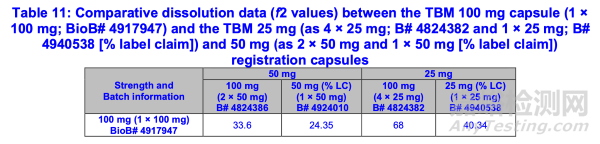
根据所提交的信息,100毫克(1×100毫克)TBM胶囊显示出与100毫克(4×25毫克)TBM胶囊相似的溶出曲线(f2>50)。然而,100毫克(2×50毫克)TBM胶囊的溶出曲线与100毫克(1×100毫克)TBM胶囊存在差异(f2<50)。FDA注意到,100毫克(1×100毫克)TBM胶囊的溶出曲线(按标示量%计)与25毫克(1×25毫克)及50毫克(1×50毫克)TBM胶囊存在差异(f2<50)。
为支持豁免TBM胶囊25毫克和50毫克规格的体内生物利用度研究,申请人在对IR#1(序列0009;附录2)的回复中申明:尽管2×50毫克注册胶囊(图2)的溶出曲线快于1×100毫克和4×25毫克注册胶囊,但2×50毫克注册胶囊的溶出曲线与2×50毫克III期胶囊相似(f2=61)——后者经证实与1×100毫克注册胶囊具有生物等效性(表10)。
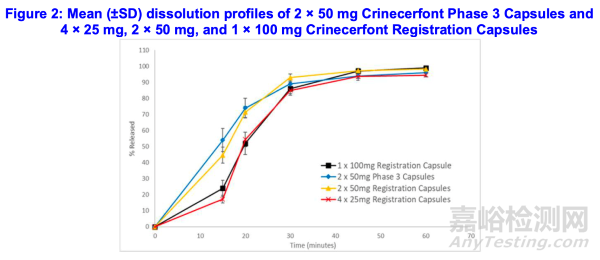
与1×100 mg和4×25 mg TBM胶囊相比(表12A和12B),2×50 mg III期和TBM胶囊观察到的更快溶出曲线可能归因于50 mg软胶囊更短的崩解时间(表12C)。
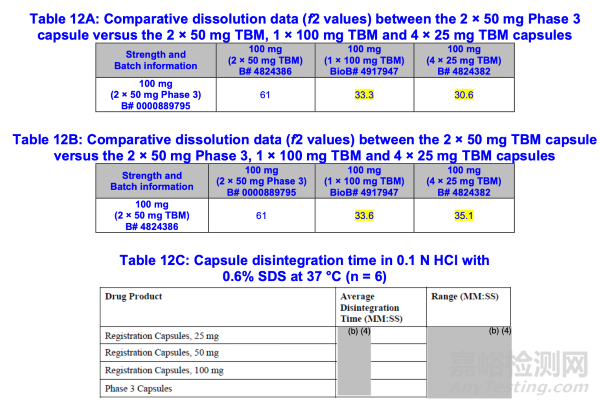
基于2×50mg III期胶囊与1×100mg注册胶囊具有生物等效性的数据(表10),以及三种规格TBM胶囊均满足"Q=45分钟内溶出%"的接受标准,FDA认为观察到的2×50mg TBM胶囊与1×100mg及4×25mg TBM胶囊溶出曲线差异,预期不会对2×50mg注册胶囊给药后制剂的体内药代动力学参数产生不良影响。
因此,基于所提交数据的整体性,本审评员认为25mg/50mg TBM制剂与100mg TBM制剂间的桥梁研究已充分建立,无需针对较低规格(25mg与50mg)进行额外的体内生物等效性研究。
FDA对比于溶出对比,详细分析了数据,归纳为:
100毫克(1×100毫克)TBM胶囊显示出与100毫克(4×25毫克)TBM胶囊有相似的溶出曲线(f2>50),但100毫克(2×50毫克)TBM胶囊的溶出曲线与100毫克(1×100毫克)TBM胶囊存在差异(f2<50)。这是同剂量条件下的溶出对比。
对于不同剂量单粒条件下,100毫克(1×100毫克)TBM胶囊的溶出曲线(按标示量%计)与25毫克(1×25毫克)及50毫克(1×50毫克)TBM胶囊存在差异(f2<50)。但2×50毫克注册胶囊的溶出曲线与2×50毫克III期胶囊相似(f2=61),后者已经证实与1×100毫克注册胶囊具有生物等效性。前面溶出不相似的原因是软硬胶囊的差异性。因此不需要再次确认注册批次的50mg规格与100mg拟定规格其体内的一致性。
生产场地与批量的桥接:
FDA评估:
采用单一场地生产了三种规格的TBM批次(附录1:表14)。TBM产品100 mg规格已开展I期研究。因此,无需进行生产场地桥接。
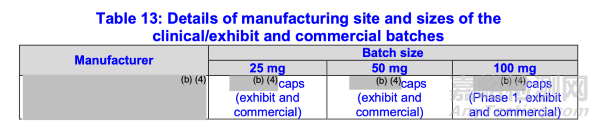
(附录1:表14)中所述三种规格TBM展示/临床批次的批量为胶囊形式。由于展示/临床批次间的批量差异在10倍范围内,基于I期研究数据桥接展示/临床批次可被接受。
拟上市三种规格商业批次的批量亦为胶囊形式。根据SUPAC-IR指南,鉴于展示/临床批次与拟上市商业批次的批量差异在10倍范围内(表13),建议通过体外溶出曲线比对桥接拟上市商业批次与临床批次。
B.13. 生物豁免请求:
FDA评估:
根据为支持不同制剂、新增规格、生产场地和批次数之间的桥接而提交的总体信息,本审查员认为依据21 CFR 320.22(d)(2)条款提交的关于TBM制剂25 mg和50 mg规格免于体内生物等效性研究的数据是可接受的。无需进行额外的体内研究。25 mg和50 mg规格的疗效研究将由临床/临床药理学审查员进行评估。
R. 区域信息:
生命周期管理考虑及风险缓解策略
FDA评估:
拟定药品为速释制剂,其达峰时间(Tmax)范围为空腹状态下7.0小时至餐后5.0小时。速释制剂出现较长达峰时间可能与活性成分crinecerfont原料药属于低溶解度/低渗透性(BCS 4类)原料药特性相关。
所申报溶出方法对关键辅料浓度变化的区分能力有限。申请人通过建立关键辅料的浓度范围及拟定/商定接受标准的方法,从生物药剂学角度将风险维持在较低水平。该风险评估基于所有适用于分装单元操作的药品生产质量管理规范均得到充分建立和持续执行的假设前提。
crinecerfont软胶囊上市了三种规格,它们的处方比例相拟,拟上市的规格之间相同剂量的溶出,50mg规格与100mg规格之间有明显的差异,f2因子小于50%。但与III期临床批次所用50mg规格对比,拟上市50mg规格的溶出与其相似,而III期临床批次所用50mg规格与III期临床所用100mg规格之间的体内等效性得到了认定,因此拟上市规格50mg其体内的等效性得到了桥接,无需再次进行体内研究。
拟上市的25mg规格,在等剂条件下与拟上市100mg规格溶出相似,因此也无需进行体内生物等效性研究,认定为已桥接。

来源:蒲公英Ouryao
关键词: CMC变更桥接评估