
嘉峪检测网 2025-05-09 09:29
导读:欧洲药品管理局于2025年3月27日发布的新指南草案专门适用于传染病mRNA疫苗,阐述了起始材料的定义,并提供了确保mRNA疫苗质量稳定所需的生产、特性、规格、分析控制和监管考虑的详细指导。
mRNA疫苗技术的快速发展,尤其得益于其在COVID-19疫情期间的成功应用,凸显了清晰、健全的监管框架的重要性。欧洲药品管理局(EMA)于2025年3月27日发布的新指南草案专门适用于传染病mRNA疫苗,包括自扩增变体,但不包括其他基于mRNA的药品。该草案与现有的欧盟法规相一致,并补充了其他相关的欧洲和ICH指南以及《欧洲药典》。
本指南阐述了起始材料的定义,并提供了确保mRNA疫苗质量稳定所需的生产、特性、规格、分析控制和监管考虑的详细指导。指南还简要介绍了菌株变化、双价和多价疫苗以及自扩增mRNA疫苗的其他监管考虑。公众意见征询截止日期为2025年9月30日。
活性物质考虑因素
该指南将mRNA本身定义为活性物质。重要的是,该指南明确了起始材料包括核苷酸、5'加帽试剂或加帽试剂以及线性DNA模板。
线性DNA模板作为起始材料
线性DNA尤其受到关注。该指南概述了提供其来源、生产工艺、注释序列以及(如适用)宿主细胞系和细胞库系统信息的必要性。
特性描述应涵盖pH值、DNA浓度、身份和序列确认、poly(A)区域完整性以及关键杂质(例如残留DNA、RNA和蛋白质)等方面。质量标准应反映工艺能力,并进行相应的论证,并提供相关的稳定性数据支持。这些要求凸显了在mRNA疫苗生产中,对线性DNA作为起始原料进行严格的定义和控制的重要性。
mRNA的表征
该指南规定了表征过程中应关注的若干关键质量属性和分析方法。具体如下表所示:
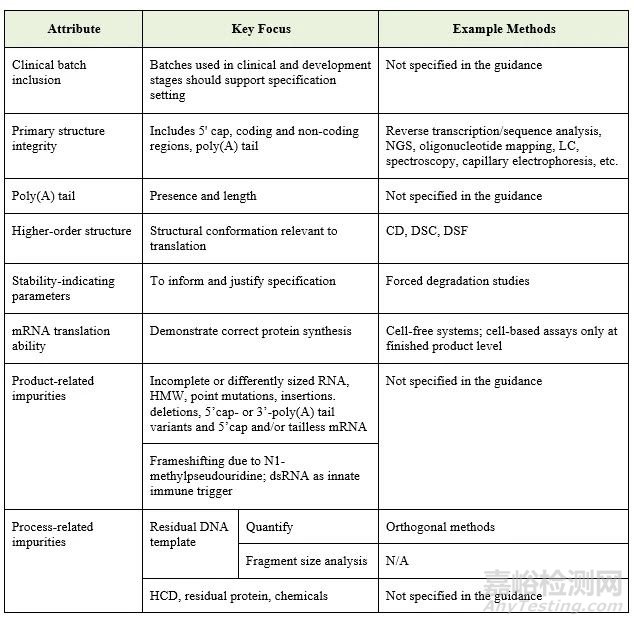
释放规格
该指南对mRNA的释放检测设定了明确的要求。规格限度应根据 ICH Q6B 进行临床验证。下表总结了药品释放时需要控制和监控的关键属性:
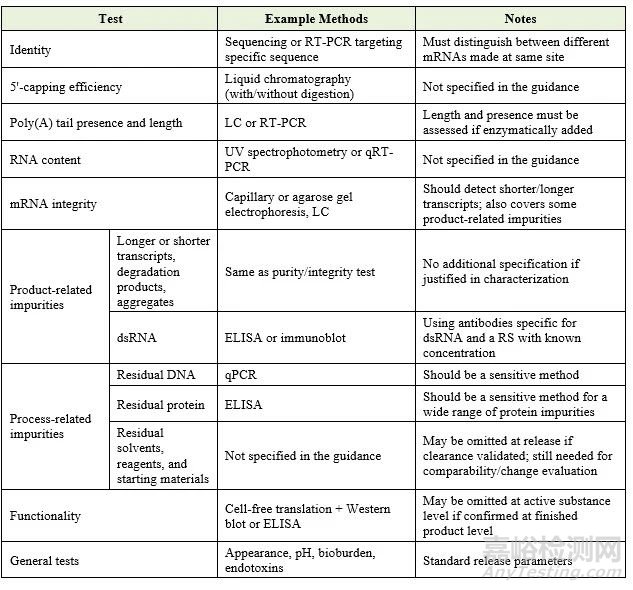
参考标准
需要时,应按照 ICH Q2(R2) 建立参考标准。
稳定性
稳定性研究应使用具有代表性的临床或商业批次来支持拟议的保质期和储存条件。关键参数通常包括mRNA完整性、poly(A)尾和RNA含量,并应反映任何中间储存温度和冻融条件。
成品考虑因素
该指南要求清楚描述剂型、容器封闭系统以及成品的完整定性和定量组成,包括所有脂质和赋形剂。
药物开发
药品开发应以明确定义的质量目标产品概况 (QTPP) 为指导,并从中得出关键质量属性 (CQA)。
赋形剂的选择,尤其是用于制备脂质纳米粒 (LNP) 的脂质,必须在安全性、功能性以及与 mRNA 的相容性方面进行论证。制剂设计应确保 mRNA 的完整性、有效包封和递送。
以下是与药物开发相关的属性列表,指示每个属性是否被标识为CQA,是否被讨论为典型的质量属性,和/或被提及为需要表征的产品或工艺相关杂质:
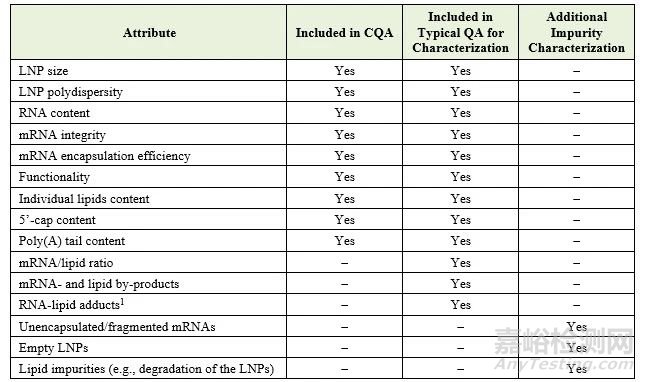
生产
制造部分应描述任何定义的中间体,为其设定适当的规格,并在适用的情况下提供其储存条件和保存时间的支持数据。
辅料的控制
辅料如有药典,应符合药典要求,并在必要时进行额外的质量属性测试,以确保其适用性。如果水是成品药品的成分,则必须符合注射用水(欧洲药典)的最低质量标准。人源性或动物源性辅料应符合 ICH Q5A 和 Q11 标准。对于新型辅料,应提供完整的生产、特性和控制信息,并附上相应的交叉引用。
成品控制
该指南概述了放行时成品测试的关键参数,如下所述。

参考标准
应根据 ICH Q6A 建立成品的参考标准,包括适用的身份、纯度和 dsRNA 定量标准。
稳定性
稳定性研究应按照 ICH Q5C 和 ICH关于新药物物质和产品稳定性测试的指导原则进行。
应监测mRNA疫苗的特定参数。如果产品解冻后在不同条件下储存,则必须提供使用稳定性支持(例如,评估冻融对完整性的影响)。稳定性测试方案应包括鉴别、纯度和功能性测试。建议的储存期应使用长期、实时、实际条件下的稳定性数据进行论证,包括在所有储存温度下进行的年度测试。保质期声明应以具有代表性的商业批次数据为依据,如有适当论证,也可使用来自生产平台的先前知识。
监管考虑
现有mRNA疫苗株的变化
在现有mRNA疫苗中引入新毒株时,指南中列出了监管机构期望的更新后的eCTD具体章节(包含详细信息)。仅可引入与新毒株相关的变更;不相关的并行变更不应一起提交。
二价和多价疫苗
对于二价和多价疫苗,指南要求对总体组成进行论证,包括每种变体的具体成分。申请人应根据生产工艺,论证检测阶段的合理性,描述关键质量属性,并提供混合物的额外特性数据。方法应能够检测和确定每种变体抗原的比例,例如使用ddPCR。
自扩增mRNA疫苗
自扩增mRNA疫苗主要在序列长度和RNA大小上存在差异,而结构特征和控制策略则相似。此外,完整的表达谱(包括复制酶和抗原)及其功能应在特性研究中验证。
替代交付系统
该指南将阳离子纳米乳剂、阳离子聚合物和阳离子肽列为替代递送系统。如果这些系统是药品成分的一部分,则应将其信息纳入3.2.P.4(eCTD)部分;如果这些系统被视为新型辅料(包括完整的描述和特性),则应将其信息纳入3.2.A.3(eCTD)部分。此类递送系统的组分也应纳入成品放行检测。
平台技术/先验知识方法
在采用基于平台技术的方法时,仍然需要针对特定产品提交相关文件。申请人有责任提供支持数据,证明先前知识的相关性,并证明拟议策略的适当性。指南指出,评估应考虑已获许可或开发的代表性产品的范围、对产品和平台特定参数的理解以及总体风险评估等因素,并将根据具体情况做出决定。
总结
EMA指南草案提炼了传染病mRNA疫苗的核心质量要求,涵盖活性物质和成品的考量,以及对菌株变化、多价制剂和基于平台方法的监管预期。该指南旨在帮助开发人员理解关键原则,并准备可靠的数据包以支持监管提交。

来源:Internet
关键词: mRNA疫苗