
嘉峪检测网 2025-05-12 13:06
导读:近日,ICH重磅发布了新的《ICH Q1稳定性试验指南》(草案),现在,国家药监局审评中心已发布其中文翻译,中英文对照如下。
近日,ICH重磅发布了新的《ICH Q1稳定性试验指南》(草案),该指南将原ICH 稳定性指南系列 Q1A-F 和Q5C进行修订并整合,增加使用过程中的稳定性研究、短期稳定性研究、中间产品/中间体加工和保持时间、标准品稳定性研究、产品生命周期和建模等内容,同时,示例了一系列产品类型的标准稳定性数据包,用于监管提交,并包括对在药品质量体系 (PQS) 内管理的研究的建议。堪称史上最强稳定性试验指南!
现在,国家药监局审评中心已发布其中文翻译,中英文对照如下:
INTERNATIONAL COUNCIL FOR HARMONISATION OF TECHNICAL REQUIREMENTS FOR PHARMACEUTICALS FOR HUMAN USE
国际人用药品注册技术协调会
ICH HARMONISED GUIDELINE
ICH 三方协调指导原则
STABILITY TESTING OF DRUG SUBSTANCES AND DRUG PRODUCTS Q1
原料药和制剂的稳定性试验(Q1)
1 INTRODUCTION
1 简介
1.1 OBJECTIVES OF THE GUIDELINE
1.1 指导原则的目的
1.2 SCOPE OF THE GUIDELINE
1.2 指导原则的适用范围
1.3 INTRODUCTION TO GUIDELINE AND GENERAL PRINCIPLES
1.3 指导原则和一般原则简介
2 DEVELOPMENT STABILITY STUDIES UNDER STRESS AND FORCED CONDITIONS
2 影响因素和强制降解条件下的开发稳定性研究
2.1 DEVELOPMENT STUDIES UNDER STRESS CONDITIONS
2.1 影响因素条件的开发研究
2.2 DEVELOPMENT STUDIES UNDER FORCED DEGRADATION CONDITIONS
2.2 强制降解条件的开发研究
2.3 ANALYSIS AND INTERPRETATION OF RESULTS
2.3 结果分析与解读
3 PROTOCOL DESIGN FOR FORMAL STABILITY STUDIES
3 正式稳定性研究的方案设计
3.1 GENERAL PRINCIPLES
3.1 一般原则
3.2 STABILITY DATA TO SUPPORT THE INITIAL RE-TEST PERIOD AND SHELF LIFE ACCORDING TO THE STANDARD APPROACH
3.2 根据标准方法支持初始复检期和有效期的稳定性数据
3.3 STABILITY-INDICATING CRITICAL QUALITY ATTRIBUTES
3.3 稳定性指示关键质量属性
3.3.1 Recommendations for Establishing a Re-Test Period or Shelf life
3.3.1 关于确定复检期 / 有效期的建议
3.3.2 Recommendations for Lifecycle Stability Protocols
3.3.2 生命周期稳定性方案建议
3.4 SPECIFICATIONS
3.4 质量标准
3.4.1 Tests and Analytical Procedures
3.4.1 测试和分析程序
3.4.2 Acceptance Criteria
3.4.2 可接受标准
3.4.3 Pharmacopoeial Critical Quality Attributes and Analytical Procedures
3.4.3 药典关键质量属性和分析方法
3.5 ADDITIONAL CONSIDERATIONS FOR VACCINES
3.5 关于疫苗的其他考虑因素
3.6 ADDITIONAL CONSIDERATIONS FOR THE COMBINATION OF A DRUG PRODUCT WITH A MEDICAL DEVICE
3.6 药械组合的其他考虑因素
3.7 RISK MANAGEMENT
3.7 风险管理
4 SELECTION OF BATCHES
4 批次选择
4.1 CONSIDERATIONS FOR SELECTION OF PRIMARY STABILITY BATCHES
4.1 注册稳定性批次选择的考虑因素
4.2 CONSIDERATIONS FOR MULTIPLE PRODUCTION SITES IN THE INITIAL REGULATORY SUBMISSION
4.2 首次注册申报中对多个生产场地的考虑因素
4.3 CONSIDERATIONS FOR VACCINES
4.3 疫苗的考虑因素
4.4 CONSIDERATIONS FOR CONTINUOUS MANUFACTURING PROCESSES
4.4 连续制造工艺的考虑因素
5 CONTAINER CLOSURE SYSTEM
5 容器密封系统
6 TESTING FREQUENCY
6 检测频率
10 SHORT - TERM STORAGE CONDITIONS
10 短期贮存条件
11 IN - USE STABILITY
11 使用中稳定性
11.1 PURPOSE OF IN - USE STABILITY TESTING
11.1 使用中稳定性试验的目的
11.2 IN - USE STABILITY STUDY PROTOCOL DESIGN
11.2.1 批次选择
11.2.1 Selection of Batches
11.2.1 批次选择
11.2.2 Selection of Analytical Procedures and Acceptance Criteria
11.2.2 使用分析方法和可接受标准的选择
11.2.3 Labelling of the In - Use Period and Storage Conditions
11.3 使用期间允许时接受贮存条件的标签
12 REFERENCE MATERIALS, NOVEL EXCIPIENTS AND ADJUVANTS
12 参比物质、新型辅料和佐剂
12.1 REFERENCE MATERIALS
12.1 参比物质
12.1.1 Considerations for Synthetic Chemical Reference Materials
12.1.1 化学药物参比物质的注意事项
12.1.2 Considerations for Biological Reference Materials
12.1.2 生物制品参比物质的注意事项
12.2 NOVEL EXCIPIENTS
12.2 新型辅料
12.3 VACCINE ADJUVANTS
12.3 疫苗佐剂
13 DATA EVALUATION
13 数据评价
13.1 GENERAL CONSIDERATIONS
13.1 一般考虑因素
13.1.1 Re - Test Period
13.1.1 复检期
13.1.2 Start of Shelf Life for Synthetic Chemical Entity Drug Products
13.1.2 化学合成实体制剂有效期起始
13.1.3 Start of Shelf Life for Biological Drug Products
13.1.3 生物制剂有效期起始
13.2 STATISTICAL EVALUATION OF THE LONG - TERM STORAGE CONDITION STABILITY PROFILE TO ESTABLISH THE RE - TEST PERIOD OR SHELF LIFE
13.2 确定复检期或有效期的长期条件稳定性特征的统计评价
13.2.1 Linear Regression for an Individual Batch
13.2.1 单一批次的线性回归
13.2.2 Combining Batches
13.2.2 合并批次
13.2.3 Scale Transformation of Data
13.2.3 数据规模转换
13.2.4 Extrapolation and Stability Modelling
13.2.4 外推和稳定性建模
13.2.5 Extrapolation for Synthetic Chemical Entities
13.2.5 化学合成实体的外推
13.2.6 No Significant Change at Accelerated Condition
13.2.6 在加速条件下无明显变化
13.2.7 Significant Change at Accelerated Condition
13.2.7 加速条件下的显著变化
13.2.8 Extrapolation for Chemical Entities when Stored Frozen
13.2.8 冷冻保存时化学实体的外推
13.2.9 Extrapolation for Biological Entities when Stored Frozen
13.2.9 生物制品外推
13.3 DATA EVALUATION FOR MULTI - FACTOR, FULL - DESIGN STUDIES
13.3 多因素、完整设计研究的数据评估
13.3.1 Testing to Combine Batch Data per Individual Combination
13.3.1 用于每个单独组合批次数据的合并测试
13.3.2 Testing to Combine Data for All Factors and Factor Combinations
13.3.2 综合所有因素和因素组合数据的测试
13.4 DATA PRESENTATION
13.4 数据呈现
14 LABELLING
14 说明书及标签
14.1 EXCURSIONS OUTSIDE OF A LABELLING CLAIM
14.1 超出说明书及标签要求的偏离
15 STABILITY CONSIDERATIONS FOR COMMITMENTS AND PRODUCT LIFECYCLE MANAGEMENT
15 关于承诺和产品生命周期管理的稳定性考虑因素
15.1 COMMITMENT STABILITY STUDIES
15.1 稳定性研究承诺
15.2 ONGOING STABILITY STUDIES
15.2 持续稳定性研究
15.3 PRODUCT LIFECYCLE STABILITY STUDIES
15.3 产品生命周期稳定性研究
15.4 STABILITY STUDIES TO SUPPORT NEW DOSAGE FORMS AND NEW STRENGTHS/CONCENTRATIONS
15.4 支持新剂型和新规格 / 浓度的稳定性研究
16 GLOSSARY
16 术语表
17 REFERENCES
17 参考文献
18 ANNEXES
18 附录
ANNEX 1 REDUCED STABILITY PROTOCOL DESIGN
附录 1 - 简化稳定性方案设计
ANNEX 2 STABILITY MODELLING
附录 2 稳定性建模
ANNEX 3 STABILITY OF ADVANCED THERAPY MEDICINAL PRODUCTS (ATMPs)
附录 3 先进治疗药品(ATMP)的稳定性
1 INTRODUCTION
1 简介
1.1 Objectives of the Guideline
1.1 指导原则的目的
The following guideline outlines the stability data expectations for drug substances and drug products.This guideline is applicable to marketed drug products, including those associated with registration andlifecycle/post-approval changes and, when applicable, master files. These applications are hereaftercollectively referred to in the guideline as regulatory submissions. ICH Q1 is a consolidated revisionthat supersedes ICH Q1A-F and Q5C guidelines and provides additional guidance on principles relatingto stability.
本指导原则概述了原料药(生物制品也称原液)和制剂的稳定性数据要求,适用于已上市产品,包括注册申报、生命周期/批准后变更相关的产品及主文件(如适用)。本指导原则统称此类申请为注册申报。ICH Q1为整合修订版,取代了ICH Q1A-F 和Q5C指导原则,并为稳定性相关原则提供了更多指导。
1.2 Scope of the Guideline
1.2 指导原则的适用范围
This guideline applies to synthetic and biological drug substances and drug products, including thefollowing:
本指导原则适用于由合成技术和生物技术制备的原料药及制剂,包括:
• Chemically synthesised drug substances including oligonucleotides, polysaccharides and polypeptides (collectively referred to as ‘synthetic chemical entities’ or ‘synthetics’ in this guideline), semi-synthetic drug substances and fermentation-derived drug substances.
• 化学合成原料药,包括寡聚核苷酸、多糖和多肽类(在本指导原则中统称为“化学合成实体”或“合成品”)、半合成原料药以及发酵原料药。•Therapeutic proteins/polypeptides, polysaccharides and proteoglycans produced usingrecombinant DNA (rDNA) technology or isolated from human, animal or plant tissues, othernatural sources, including body fluids (such as plasma-derived products), or cell cultures.
• 采用重组DNA(rDNA)技术生产或从人体、动物或植物组织,其他天然来源(包
括体液,如血浆衍生产品)或细胞培养物中分离的治疗性蛋白/多肽、多糖和蛋白聚糖。
• Conjugated products that are made up of proteins/polypeptides linked to another moiety (e.g.,antibody-drug conjugate).
• 由蛋白/多肽与其他分子偶联形成的制品(如抗体药物偶联物)。
• Vaccines, allergenic products, and adjuvants.
• 疫苗、过敏原产品和佐剂。
• Autologous and allogenic cell-based substances, including those which may be geneticallymodified ex-vivo (refer to Annex 3 – Stability of Advanced Therapy Medicinal Products(ATMPs)).
• 自体和异体细胞类物质,包括体外基因修饰产品(见附录3-先进治疗药品(ATMP)
的稳定性)。
• Gene therapy products that mediate their effect by the expression (transcription or translation)of transferred genetic materials and genome editing products used to modify cells (refer toAnnex 3 – Stability of Advanced Therapy Medicinal Products (ATMPs)).
• 通过遗传物质的表达(转录或翻译)介导其作用的基因治疗产品,以及用于细胞修
饰的基因编辑产品(见附录3-先进治疗药品(ATMP)的稳定性)。
• The drug constituent part of a combination of a drug product with a medical device (both integral or co-packaged).
• 药械组合(单一实体或组合包装)的药物组成部分。
• Co-packaged solvents/diluents.
• 组合包装的溶剂/稀释剂。
• Natural health products that are regulated as drug products.
• 按药品监管的天然保健品。
The guideline is applicable to all regulatory submissions and, in accordance with regional regulations,can apply to prescription and non-prescription drug products (e.g., regulated over-the-counter products),original drug products (e.g., new entities), new product presentations, abbreviated/abridged applications(e.g., generics) and biosimilars.
本指导原则适用于所有注册申报,同时根据地区法规,可适用于处方药品和非处方药品(如受监管的非处方药品)、原研药品(如新化学实体)、新剂型、简化申请(如仿制药)及生物类似药。
The principles outlined in this guideline are applicable to support post-approval changes (PACs) thatrequire supportive and confirmatory stability studies, including those that are discussed within ICHQ12.
本指导原则适用于需要进行支持性和确证性稳定性研究的批准后变更(PAC),包括ICH
Q12中讨论的情形。
Although this guideline is not directly applicable to drug substances and drug products during clinicaldevelopment stages, the concepts can apply proportionate to increasing level of product and processunderstanding during pharmaceutical development. The data from development batches that meetprimary stability requirements may be used to support a regulatory submission and for product lifecyclemanagement. Refer to Section 15 - Stability Considerations for Commitments and Product LifecyleManagement.
本指导原则虽然不直接适用于临床开发阶段的原料药和制剂,但随着药学开发中对产品和工艺的认知加深可逐渐适用。符合的开发批次的数据可用于支持注册申报及产品生命周期管理。见第15节“稳定性承诺和产品生命周期管理的考量”)。
The guideline is not applicable to device constituent parts, radiopharmaceuticals and whole bloodproducts.
本指导原则不适用于器械组成部分、放射性药物和全血制品。
1.3 Introduction to Guideline and General Principles
1.3 指导原则和一般原则简介
The purpose of stability testing is to provide evidence on how the quality of a drug substance or drugproduct varies with time under the influence of a variety of environmental and physical factors such astemperature, humidity, light, or agitation. Stability testing establishes and confirms a re-test period orshelf life for the drug substance or a shelf life for the drug product in the proposed container closuresystem under the recommended storage conditions. Shelf life is also referred to as dating period orexpiry period in some regions. This guideline provides comprehensive guidance to establish stabilityfor all molecule types within its scope and includes recommendations on how science- and risk-basedprinciples may be applied. A standard approach to assess each stability-related topic is provided bydescribing the general principles and strategies to assess stability. In addition, the principles of Qualityby Design described within ICH Q8-Q11 and Q14, through enhanced understanding of critical quality attributes (CQAs) and the impact that the manufacturing process can have on these attributes, areapplicable to the design of an overall stability strategy.
稳定性试验的目的在于提供证据,以说明在温度、湿度、光照或搅拌等各种环境因素和物理因素的影响下,原料药或制剂的质量如何随时间变化。稳定性研究旨在建立并确认在拟定贮藏条件下及拟定的包装系统中原料药的复检期或有效期,或制剂的有效期。某些地区,有效期(Shelf life)也称为有效日期(dating period)或失效日期(expiry period)。本指导原则为其适用范围内的所有分子类型提供全面的稳定性研究指导,并就如何应用基于科学和风险的原则提出了建议。通过阐述稳定性评估的一般原则和策略,本指导原则提供了评估各稳定性相关议题的标准方法。此外,ICH Q8-Q11和Q14综述了质量源于设计的原则,因其对关键质量属性(CQAs)以及生产工艺对这些属性影响的深入理解,可将其应用于整体稳定性策略的设计。
This guideline should be considered in its entirety for a comprehensive approach to stability studies.
在制定稳定性研究的综合方法时,应全面考虑本指导原则。
The guideline exemplifies the standard stability data package for drug substances and drug productsand provides guidance on alternative and scientifically justified approaches that encompass the varietyof different situations that may be encountered due to specific scientific considerations and characteristics of the data being evaluated. Alternative strategies based on science- and risk-based principles (e.g., as described in ICH Q8-Q11 and section IX of ICH Q12) for drug substances and drug products may be proposed by the applicant of a regulatory submission, leveraging quality risk management principles, pharmaceutical development data (e.g., as discussed in Section 2 – Development Studies Under Stressed and Forced Conditions), prior knowledge and modelling, (e.g., as discussed in Annex 2 -Stability Modelling). Examples are provided under specific sections to illustrate how science- and risk-based strategies may be applied.
本指导原则通过示例形式,说明了原料药和制剂的标准稳定性数据包,并为替代性及科学论证的方法提供指导,这些方法涵盖了因被评估数据的特定科学考量和特性而可能遇到的各种不同情况。注册申报的申请人可基于对原料药和制剂的科学和风险原则(如ICH Q8-Q11和ICH Q12 的IX部分所述),利用质量风险管理原则、产品开发数据(如第2节“影响因素条件和强制降解条件下的开发研究”所述)、先验知识和建模,(如附录2-稳定性建模所述),提出替代策略。具体章节提供的示例说明了如何应用基于科学和风险的策略。
Unless otherwise specified, the recommendations described in this guideline apply to both drug substance and drug products. Additionally:
除非另有说明,本指导原则中的建议同时适用于原料药和制剂。此外:
• Each section may include guidance for specific product types (e.g., synthetics, biologicals,vaccines or a combination drug product with a medical device) where relevant.
• 各章节在适用时可包含针对特定产品类型的指导(如,合成药物、生物制品、疫苗
或药械组合)。
• For semi-synthetics, fermentation and conjugated products, the recommendations for syntheticsand biologicals would apply, as appropriate.
• 对于半合成、发酵和偶联产品,酌情适用合成药物和生物制品的建议。
• Where “products” is mentioned by itself in this guideline, this is to be interpreted as “drugsubstances and drug products”.
• 本指导原则中提及的“产品”,应理解为“原料药和制剂”。
• Recommendations on the general principles for stability studies and data expectations for drugsubstances and drug products apply across all climatic zones for regulatory submissions andlifecycle management. The mean kinetic temperature in any part of the world can be derivedfrom climatic data, which divides the world into four climatic zones, I-IV (13, 14). The fourzones are distinguished by their characteristic prevalent annual climatic conditions based on theconcept originally described by W. Grimm (15), updated in W. Grimm (16) and adopted underWHO Technical Reports (13, 14). This guideline addresses all four climatic zones. Theprinciple has been established that if the stability information is generated under a more severeclimatic zone storage condition, it would be acceptable in the other climatic zones, providedthe information is consistent with this guideline and the labelling and storage statements are inaccordance with regional requirements.
• 关于原料药和制剂稳定性研究的一般原则及数据要求的建议,适用于所有气候带的注册申报与生命周期管理。基于气候数据可推导全球任何地区的平均动力学温度,据此将全球划分为I-IV四个气候带(13、14)。四个气候带的划分基于W.Grimm(15)最初提出的理论框架,该分类依据各地区显著的全年普遍气候条件特征,后续在W.Grimm(16)的更新研究中得到完善,并被WHO技术报告(13、14)所采纳。
本指导原则涵盖全部四个气候带,确定如下原则:若稳定性信息是在更严苛的气候带贮藏条件下生成,且符合本指导原则要求,同时说明书及标签的贮藏声明符合地区要求,则可接受用于其他气候带。
• The recommendations may be applicable to drug substance intermediates and drug productintermediates. Intermediates that are stored as part of manufacturing process activities (e.g.,unprocessed bulk harvest, granulations) should be evaluated in accordance with Section 9 -Stability Considerations for Processing and Holding Times for Intermediates. For those
intermediates that are packaged and stored outside of manufacturing process activities, a
holding time may be established or it may be appropriate to establish a re-test period or shelf
life as per the applicable sections of this guideline (e.g., antibody prior to conjugation and a
spray dried dispersion).
• 本指导原则的建议可适用于原料药中间产品和制剂中间产品。作为生产工艺活动(如未加工的原液、颗粒混合物)组成部分贮藏的中间产品,应根据第9节“中间产品加工和保持时限的稳定性考量”进行评估。对于在生产工艺活动外进行包装和贮藏的中间产品,可依据本指导原则的适用章节(如偶联前的抗体和喷雾干燥分散体)建立保持时限,或适当设定复检期或有效期。
• The recommendations may be applicable to reference materials as well as to drug productscontaining certain excipients and adjuvants where the stability of these components cansignificantly impact drug product performance. Refer to Section 12- Reference Materials,
Novel Excipients and Adjuvants for detailed guidance. Co-packaged solvents/diluents should
follow the recommendations for drug products.
• 本指导原则的建议可适用于标准物质,以及含有特定辅料与佐剂的制剂,其成分的
稳定性可能显著影响制剂的性能。具体指导见第12节“标准物质、新型辅料与佐剂”。
组合包装的溶剂/稀释剂应遵循制剂的相关建议。
• Regulatory expectations for the stability data package in this guideline are also applicable todrug substances and drug products made using continuous manufacturing (CM) processes.
• 本指导原则对稳定性数据包的监管要求同样适用于通过连续制造(CM)工艺生产
的原料药和制剂。
• Annexes are intended to either supplement the guideline with specific guidance on enhancedapproaches or to provide product-specific guidance for product types with specific and uniquestability considerations. Annex 1 provides guidance on Reduced Protocol Design; Annex 2provides guidance on Stability Modelling; and Annex 3 provides Additional Considerations forATMPs.
The main types of stability studies are graphically represented in Figure 1.
• 附录旨在通过以下方式补充本指导原则:提供针对强化方法的具体指导,或为具有
特殊和独特稳定性考量的产品类型提供专项指导。附录1提供了简化方案设计的指导;附录2提供了稳定性建模的指导;附录3提供了先进治疗产品(ATMP)的其他考量。
稳定性研究的主要类型如图1所示。
Figure 1: Stability Study Types
图1 稳定性研究类型
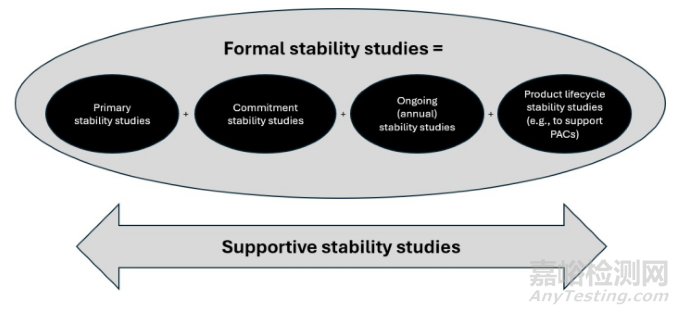
Formal stability studies are primary, commitment, ongoing and product lifecycle stability studies conducted under the accelerated, intermediate, or long-term storage conditions (as applicable) to establish or confirm a re-test period or a shelf life. Supportive stability studies are those stability studies that are conducted (as applicable) to support the practical use of the product (including label claims) or a re-test period or a shelf life (e.g., photostability, in-use, short-term storage condition studies and studies to support excursions or modelling). Formal and supportive stability studies and their purposes are described in various sections of this guideline. In addition to formal stability studies, guidance is provided on studies that inform stability knowledge and product understanding (refer to Section 2 – Development Studies Under Stressed and Forced Conditions).These development studies are introduced in Section 2 because some of this information is utilised to develop the primary stability protocol and the validation of stability-indicating methodologies.
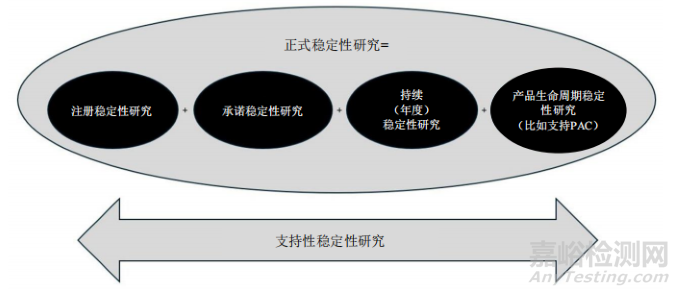
正式稳定性研究系指在加速条件、中间条件和长期条件下(如适用)开展的所有注册、承诺、持续或产品生命周期稳定性研究,以建立或确认复检期或有效期。支持性稳定性研究系指为支持产品的实际使用(包括说明书及标签声明)或复检期或有效期而开展的稳定性研究(如适用)(如光稳定性、使用期间、短期贮存条件研究和支持偏离或建模的研究)。本指导原则各章节对正式与支持性稳定性研究及其目的进行了阐述。除正式稳定性研究外,本指导原则还提供增强稳定性认知与产品理解的研究指导(见第2节“影响因素条件和强制降解条件下的开发研究”)。第2节对此类开发研究进行了介绍,因其中部分信息用于制定注册稳定性方案和验证稳定性的指示方法。
The guideline discusses strategies for protocol design within Section 3 - Stability Protocol Design to Section 7 - Storage Conditions. The recommendations in these sections are applicable to primary stability studies. However, the principles of protocol design are intended to apply to any stability protocol (e.g., commitment, ongoing and product lifecycle stability studies, including those to support changes).
本指导原则在第3节“稳定性方案设计”至第7节“贮藏条件”讨论了方案设计策略。上述章节的建议适用于注册稳定性研究。但方案设计的原则拟适用于所有稳定性方案(如承诺、持续和产品生命周期稳定性研究,以及变更支持性研究)。
The concept of a ‘representative batch’ to support establishing the re-test period or shelf life is referenced throughout this guideline. The justification that a batch is representative will vary depending on the drug substance and drug product types, their complexity and manufacturing processes. This is discussed in detail within Section 4 - Selection of Batches.
本指导原则全文引用了“代表性批次”概念以支持确定复检期或有效期。批次具有代表性的论证因原料药和制剂的类型、复杂程度和生产工艺而异,详见第4节“批次选择”。
The applicant should consider all available stability knowledge when designing stability protocols and defining information for inclusion on the product labelling (e.g., storage statements). This includes considerations of the impact of holding times, the primary stability data and supportive stability data to inform long-term, short-term and in-use storage conditions. In many cases, stability protocol designs may be dependent on the potential impact on the final product quality and therefore based on quality risk management.
申请人在设计稳定性方案和制定产品说明书及标签信息(如贮藏声明)时,应综合考虑所有可用的稳定性知识,包括保持时限的影响、注册稳定性数据和支持性稳定性数据对长期、短期和使用期间的贮藏条件。多数情况下,稳定性方案的设计可能取决于对终产品质量的潜在影响,因此应基于质量风险管理原则。
This guideline does not specify filing mechanisms or regional requirements.
本指导原则不涉及具体的申报机制或区域要求。
2 DEVELOPMENT STABILITY STUDIES UNDER STRESS AND FORCED
CONDITIONS
2 影响因素和强制降解条件下的开发稳定性研究
Product knowledge is useful in the design of formal stability study protocols. Development studies may be useful to characterise the physical, chemical and biological changes likely to occur with storage, to establish the degradation profile and intrinsic stability of the product, to confirm and validate the stability-indicating nature of the analytical procedures, to inform specifications and to determine whether unexpected exposures to conditions other than those defined in the label are deleterious to the product (refer to Section 14 – Excursions Outside of a Labelling Claim). In addition, these development studies can be used to help design the primary stability protocol and may also be applied to protocols used to support changes during the product lifecycle (refer to Section 3 - Stability Protocol Design and Section 15 - Stability Considerations for Commitments and Product Lifecyle Management).
产品认知有助于设计正式稳定性研究的方案。开发研究可能有助于表征贮藏期间可能发生的潜在的物理、化学和生物特性变化,以建立产品的降解特性谱和固有稳定性,确认并验证分析方法的稳定性指示能力,作为制定质量标准的依据,并评估偏离说明书及标签贮藏条件对产品的危害(见第14节“超出说明书及标签声明的偏离”)。此外,开发研究可用于帮助设计注册稳定性方案,也可应用于支持产品生命周期变更的方案(见第3节“稳定性方案设计”和第15节“稳定性承诺和产品生命周期管理的稳定性考量”)。
In the context of generating product knowledge, studies may be performed under accelerated and/or stress conditions, including forced conditions. The nature of this testing should be proportionate to the knowledge available, the type of the drug substance or drug product being evaluated and the quality attribute(s) being investigated.
已有产品认知的前提下,可以在加速和/或影响因素条件下(包括强制降解条件)进行研究。试验的性质应与现有认知水平、所评价原料药或制剂的类型以及所研究的质量属性相称。
Accelerated conditions (temperature and when applicable, humidity), over a defined time period, are intended to increase the rate of chemical degradation, physical change and/or biochemical change in the product. Data generated under accelerated conditions can be used to gain product knowledge and to support extrapolation, re-test or shelf life determination and to evaluate the impact of excursions outside the label storage conditions. Accelerated testing is typically included as part of the formal stability program as described in Sections 3 – Stability Protocol Design through Section 7 – Storage Conditions.
在规定时间内,加速条件(温度和湿度,如适用)旨在提高产品的化学降解、物理变化和/或生化反应的速率。加速条件下的数据可用于获取产品认知,支持外推、复检期或有效期的确定,并评估偏离说明书及标签贮藏条件的影响。如第3节“稳定性方案设计”至第7节“贮藏条件”所述,加速试验通常作为正式稳定性计划的一部分。
Development studies undertaken to assess the effect of stress on the drug substance and/or drug product can be divided into two categories:
为评估影响因素条件对原料药和/或制剂的影响进行的开发研究可分为两类:
1) Studies conducted under stress conditions: Conditions are more severe than the accelerated conditions but not necessarily intended to deliberately degrade the sample.
1) 影响因素条件研究:条件比加速条件严苛,但非刻意诱导样品降解。
2) Studies conducted under forced degradation conditions: Conditions are intended to deliberately degrade the sample (such as elevated temperature, humidity, pH, oxidation, agitation and light).
2) 强制降解研究:刻意诱导样品降解(如高温、高湿、pH值、氧化、搅拌和光照)。
The purpose of this section is to describe the principles of development studies under stress and forced conditions. This section provides clarity on the concepts, study design and considerations for interpreting the results.
本节旨在介绍影响因素条件和强制降解条件下开发研究的基本原则。阐明了概念、研究设计和结果解释的考虑因素。
2.1 Development Studies Under Stress Conditions
2.1 影响因素条件的开发研究
Studies under stress conditions can contribute to an understanding of product knowledge and the data gathered from these studies can be useful in addressing unexpected excursions outside of the conditions defined on the labelling (refer to Section 14.1 – Excursions Outside of a Labelling Claim).
影响因素条件研究有助于增进产品认知,其数据可用于应对超出说明书及标签规定条件的意外偏离(见第14.1节“超出说明书及标签声明的偏离”)。
Stress condition studies can include temperature and humidity levels above accelerated conditions,thermal cycling and freeze-thaw studies, as appropriate. For synthetic chemicals entities, these studies may be conducted on one batch of the drug product and where relevant one batch of the drug substance directly exposed or in a container closure system, as applicable. For biologicals, at a minimum, stress studies may be performed on a single batch of drug product, however, it may be possible to justify using a single batch of drug substance if it is representative of the drug product.
影响因素条件研究可以包括高于加速条件的温度和湿度水平、热循环和冻融试验(如适用)。对于化学合成实体,上述研究可采用一批制剂进行(必要时可同时采用一批原料药进行,原料药可采用直接暴露方式或置于容器密封系统,如适用)。对于生物制品,至少应对一批制剂进行影响因素研究,但如果原料药批次能代表制剂特性,则可以提供合理性论证后使用该批原料药。
2.2 Development Studies Under Forced Degradation Conditions
2.2 强制降解条件的开发研究
Forced degradation studies may be utilised to investigate potential degradation pathways; gain product knowledge; understand the intrinsic stability of product and used to develop and confirm stability-indicating nature of the analytical procedure (refer to ICH Q2 and ICH Q14). It is acceptable to leverage product knowledge when data is available on identified degradation products and pathways, including scientific literature.
强制降解研究可用于研究潜在的降解途径;获取产品知识;了解产品的固有稳定性,并用于开发和确认分析方法具有稳定性指示特性(见ICH Q2和ICH Q14)。当可获得已鉴定的降解产物和降解途径的数据时(包括科学文献),可以利用现有产品知识。
It is recommended to assess forced conditions on a single batch of the drug substance. It should include the effect of elevated temperatures, humidity (e.g., 75% Relative Humidity (RH) or greater) where appropriate, oxidation and photodegradation on the drug substance. Testing should evaluate the susceptibility of the drug substance to hydrolysis across a range of pH values. Also, a combination of forced conditions may be appropriate to test under certain circumstances (e.g., agitation and heat).
建议对单批原料药的强制降解条件进行评估,评估应包括温度、湿度(如≥75% 相对湿度(RH),如适用)、氧化和光降解的影响。应评估原料药在一定pH值范围内对水解的敏感性。此外,在某些情况下(如搅拌和加热),可能需要组合使用强制降解条件进行试验。
For drug products, testing under forced conditions is recommended on a single batch of exposed drug product. It should include the effect of temperature, humidity (e.g., 75% RH or greater) where appropriate and light. Additional forced conditions for specific types of products and dosage forms may be appropriate.
对于制剂,建议对单批制剂开展强制降解条件,试验应包括温度、湿度(如≥75% RH,如适用)和光照的影响。对于特定类型的产品和剂型,可能需要进行额外的强制降解条件。
For biologicals, studies under forced degradation conditions should be performed on a single batch of drug substance; alternatively, it may be possible to justify using a single batch of drug product.
对于生物制品,应对单批原料药进行强制降解条件的研究,或者,如果能证明制剂批次具有代表性,也可使用单批制剂。
The forced photodegradation condition can be an integral part of forced degradation studies. The purpose of forced photodegradation studies is to evaluate the overall photosensitivity of the product. A forced photodegradation study requires exposure to light conditions which are more extreme than the light conditions utilised in confirmatory studies (refer to Section 8 – Photostability).
强制光降解条件可作为强制降解研究的组成部分,其目的是评估产品的整体光敏性。强制光降解研究需要采用比确证性研究更严苛的光照条件(见第8节“光稳定性”)。
With forced degradation studies, the conditions and duration may need to be varied depending on the sensitivity of the product. For development and analytical procedure validation purposes, it is appropriate to limit the exposure and end the forced degradation study if extensive decomposition occurs. Similarly, for stable materials, studies may be terminated after an appropriate exposure level has been used. The design of these experiments is left to the applicant’s discretion although the exposure levels used should be justified.
强制降解研究的条件和持续时间可根据产品的敏感性进行调整。开发和分析方法验证时,若发生显著性的分解,则可适当限制暴露并终止研究。同样地,对于稳定的样品,在达到适当暴露水平后可终止研究。申请人可自主设计试验方案,但应对所用的暴露水平进行论证。
2.3 Analysis and Interpretation of Results
2.3 结果分析与解读
When testing under stressed conditions, including forced degradation, samples should be examined at the end of the exposure period for any changes in physical, chemical, or biological properties (e.g., physical state, clarity, colour, degradation products, particle size, potency), as applicable, by a procedure suitable to detect any evidence of change.
在影响因素条件下(包括强制降解)进行试验时,应采用适当的检测方法,在暴露期结束时评估样品的物理、化学或生物学特性的变化(如物理状态、澄清度、颜色、降解产物、粒度、效价)(如适用)。
Changes in attributes that are unlikely to occur under normal storage conditions may occur under forced conditions and possibly under stress conditions (e.g., the formation of degradation products). This information may be useful in developing and validating suitable analytical procedures and can be part of a comprehensive approach to justify the overall control strategy.
在常规贮藏条件下不太可能发生的属性变化,但在强制降解条件和影响因素条件下可能发生(如形成降解产物)。这些信息有助于开发和验证分析方法,并可作为综合方法的一部分,用以证明整体控制策略的合理性。
The data obtained from these development studies may also inform product understanding and help identify the potential stability-indicating CQAs that should be monitored during stability testing,assisting in the design of the stability protocol (refer to Section 3 - Stability Protocol Design). Although forced degradation studies are not part of the formal stability studies, results from the forced degradation studies are an integral part of the information provided to regulatory authorities (e.g., support analytical procedure validation, product characterisation, specifications or packaging considerations). Data from development studies under stress condition should be included in regulatory submissions if they support a claim on the product labelling.
这些开发研究数据可增进产品认知,帮助识别稳定性研究中需监测的稳定性指示CQA,从而协助设计稳定性方案(见第3节“稳定性方案设计”)。虽然强制降解研究不属于正式稳定性研究,但其结果是提交给监管机构的重要组成部分(如支持分析方法验证、产品表征、质量标准或包装的筛选)。若影响因素条件下的开发研究数据支持药品说明书及标签的声明,则应将其纳入注册申报资料中。
3 PROTOCOL DESIGN FOR FORMAL STABILITY STUDIES
3 正式稳定性研究的方案设计
This section provides guidance that is intended to be used in conjunction with Section 4 – Selection of Batches through Section 7 – Storage Conditions to establish a formal stability study protocol. Figure 2 illustrates how an applicant may approach the design and development of a formal stability protocol.The “available stability data” in the figure refers to knowledge gained from long-term and accelerated stability studies conducted earlier in development and from development studies discussed in Section 2 – Development Studies Conducted on Stressed and Forced Conditions.
本节提供了旨在与第4节“批次选择”至第7节“贮藏条件”配合使用的指导,以建立正式稳定性研究的方案。图2说明了申请人如何设计和制定正式稳定性研究的方案。图中的“现有稳定性数据”是指从开发早期进行的长期和加速稳定性研究以及第2节“强力条件和强制降解条件下的开发研究”中获得的知识。
Where noted, these sections provide specific guidance for establishing a primary stability protocol to determine a re-test period or shelf life (refer to Section 13 - Data Evaluation). When applicable, the guidance in these sections should be utilised in conjunction with Section 15 - Stability Considerations for Commitments and Product Lifecyle Management (for commitment stability studies, ongoing stability studies and lifecycle stability studies) and Annex 1 - Reduced Stability Protocol Design (where reduced study designs may be appropriate).
如有注明,这些章节提供了关于制定注册稳定性方案的具体指导,以确定复检期/有效期(见第13节“数据评估”)。在适用的情况下,这些章节中的指导应与第15节“关于承诺与产品生命周期管理的稳定性考虑因素”(适用于承诺稳定性研究、持续稳定性研究和生命周期稳定性研究)和附录1-简化稳定性方案设计(可能适合简化研究设计)配合使用。
3.1 General Principles
3.1 一般原则
A summary of the stability protocol should be provided in a regulatory submission when a re-test period or shelf life is to be established or confirmed. The stability protocol incorporates all necessary information to establish or confirm the stability of the drug substance or drug product under the recommended storage conditions throughout the re-test period or shelf life. This includes consideration of data from primary stability studies and supporting data to inform long-term storage, short-term storage, excursions and in-use conditions.
当需要建立或确认复检期/有效期时,应在注册申报资料中纳入稳定性方案的摘要。稳定性方案应包含所有必要的信息,以确认原料药或制剂在其整个复检期/有效期内在推荐的贮藏条件下的稳定性。这包括对注册稳定性研究的数据和支持性数据的考虑,以告知长期贮藏、短期贮藏、偏离和使用期间条件。
An illustration of the general process for the development, design and execution of a stability protocol is shown in Figure 2. The applicant is responsible for building knowledge and understanding during pharmaceutical development, leading to the identification of those CQAs that are or have the potential to be stability-indicating under appropriate storage conditions and using this information to design the protocol to support the formal stability studies. Stability studies should include testing of those attributes that are susceptible to change during storage and can potentially influence quality, safety and efficacy.During the product’s lifecycle, as knowledge is gained, stability protocol designs may be optimised. Changes to the stability protocol to extend a re-test period or shelf life should be established in accordance with Section 15 - Stability Considerations for Commitments and Product Lifecyle Management.
稳定性方案的开发、设计和执行的一般过程如图2所示。申请人负责在产品开发过程中积累知识和理解,从而确定在适当贮藏条件下具有或可能指示稳定性的CQA,并使用该信息设计方案,以支持正式稳定性研究。稳定性研究应包括对贮藏期间易发生变化且可能影响质量、安全性和有效性的属性进行检测。在产品的生命周期中,随着知识的不断获取,可以不断优化稳定性方案设计。为延长复检期/有效期而对稳定性方案进行的变更,应根据第15节“关于承诺和产品生命周期管理的稳定性考虑因素”进行确定。
Figure 2: General Process Flow for the Development, Design and Execution of a Stability Protocol
图2 稳定性方案开发、设计和执行的一般流程
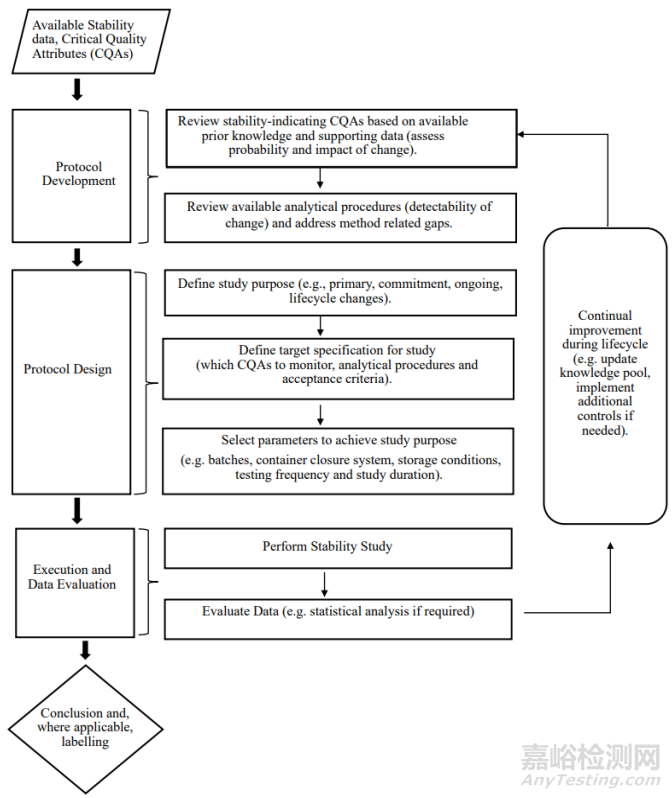
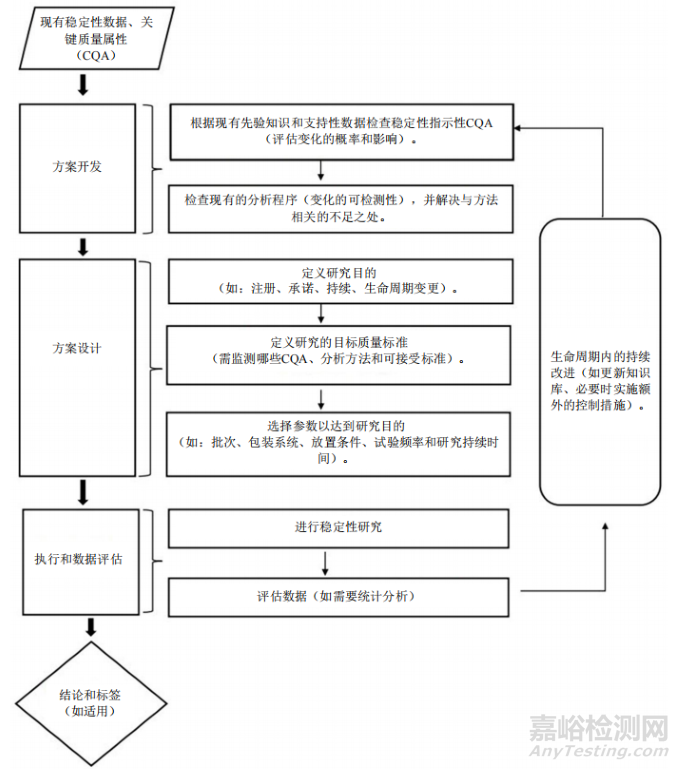
The principles detailed for protocol design should be applied from initial regulatory submission through product lifecycle. The precise protocol design will depend on the drug substance/drug product, study purpose and the available prior knowledge.
方案设计的详细原则应涵盖从首次注册申报到产品生命周期的所有内容。方案设计精确与否将取决于原料药/制剂、研究目的和可用的先验知识。
Additional protocol considerations for photostability, excursions, short-term storage and in-use conditions are described in the respective sections (refer to Section 8 – Photostability, Section 14.1 –Excursions Outside of a Labelling Claim, Section 10 - Short-Term Storage Conditions and Section 11– In-Use Stability).
关于光稳定性、偏离、短期贮藏和使用期间条件的其他方案考虑因素,在相应章节中均有描述(见第8节“光稳定性”、第14.1节“超出说明书及标签声明的偏离”、第10节“短期贮存条件”和第11节“使用中稳定性”)。
A full design stability protocol is a protocol where at least three batches of the drug substance or at least three batches of each strength of the drug product covering the proposed container closure systems for every combination of all design factors are included and tested at all time points. Alternative approaches to stability protocol design, such as bracketing, matrixing, knowledge- and risk-based protocol reductions and stability models are described in Annex 1 – Reduced Stability Protocol Design and Annex 2 – Stability Modelling. Additional considerations for ATMPs are provided in Annex 3 – Stability of Advanced Therapy Medicinal Products (ATMPs).
完整稳定性设计方案是指在所有时间点纳入并检测至少三批原料药或每种规格至少三批制剂(涵盖所有设计因素的每种组合的拟定容器密封系统)的方案。稳定性方案设计的替代方案,如括号法、矩阵设计、基于知识和风险的方案简略以及稳定性模型,见附录1-简化稳定性方案设计和附录2-稳定性建模。附录3-先进治疗产品(ATMP)的稳定性中介绍了ATMP的其他考虑因素。
3.2 Stability Data to Support the Initial Re-test Period and Shelf Life According to the
Standard Approach
3.2 根据标准方法支持初始复检期和有效期的稳定性数据
This section provides guidance on establishing the re-test period, shelf life and storage conditions using data from the primary stability study (refer to Section 4 – Selection of Batches). This is considered the standard approach. When the standard approach is adopted, the recommendations provided in Table 1 establish an appropriate minimum dataset at the time of the initial regulatory submission to assign a re- test period and shelf life in accordance with the guidance provided in Section 13 – Data Evaluation.Alternative approaches to the principles and practices described in this section may be acceptable if they are supported by adequate justification, including an enhanced knowledge of product performance from prior knowledge, as per ICH Q8 - Q11 and modelling as discussed in Annex 2 - Stability Modelling.
本节对使用注册稳定性研究数据确定复检期、有效期和贮藏条件提供了指导(见第4节“批次选择”)。这被认为是标准方法。当采用标准方法时,表1中的建议是在首次申报时建立一个适当的最小数据集,以便根据第13节“数据评估”中提供的指导确定复检期/有效期。如果有充分的依据支持,包括根据ICH Q8-Q11和附录2-稳定性建模中讨论的建模,从先验知识中增强对产品性能的了解,则可以使用本节所述原则和实践的替代方案。
The stability package provided in the regulatory submission should be sufficient to support the proposed re-test period or shelf life and storage conditions. The long-term stability protocol should, at a minimum, ensure testing continues for the duration of the proposed re-test period or shelf life.
注册申报资料中提供的稳定性数据包应足以支持拟定的复检期/有效期和贮藏条件。长期稳定性方案至少应确保检测在建议的复检期/有效期内持续进行。
Data from the accelerated storage conditions and, if appropriate, from the intermediate storage conditions can be used to evaluate the effect of short-term excursions outside the labelled storage conditions (e.g., during shipping). For synthetics, data from the accelerated storage condition are also needed to enable extrapolation in accordance with Section 13.2.5 – Extrapolation for Synthetic Chemical Entities. For biologicals, data from the accelerated storage condition is utilised for product understanding and may be used to support analytical comparability. Even though data generated under accelerated storage conditions are not used to establish a re-test period or shelf life for biologicals, it is strongly suggested to include these data in the regulatory submission.
来自加速条件和中间条件(如适用)的数据可用于评估短期偏离标签上的贮藏条件(比如运输期间)的影响。对于化学合成实体,还需要加速条件下的数据,以便根据第13.2.5节“化学合成实体的外推法”进行外推。对于生物制品,加速条件下的数据用于产品理解,并可用于支持分析可比性。尽管加速条件下生成的数据不用于建立生物制品的复检期/有效期,但本指导原则强烈建议将这些数据纳入注册申报资料中。
Refer to Section 4 – Selection of Batches, Table 2 for guidance on selection of primary batches. For synthetics and for biologicals, a primary batch may be a production batch but does not need to be a production batch.
有关注册稳定性批次选择的指导,请参阅第4节“批次选择”中的表2。对于化学合成实体和生物制品,注册稳定性批次可以是生产批次,但不必须是生产批次。
Biological drug substances and drug products usually require stringent conditions for their storage to ensure maintenance of biological activity and to avoid degradation, because of dependence of molecular conformation and biological activity on noncovalent as well as covalent forces, resulting their high sensitivity to environmental factors (e.g., temperature changes, oxidation, light, ionic content and shear). The evaluation of their stability may necessitate complex analytical methodologies including physicochemical, biochemical and immunochemical methods, and consideration of many external conditions which can affect the product’s potency, purity and quality. For biological drug substances and drug products, data from three primary batches that cover the duration of the proposed shelf life should be submitted unless an alternative approach is justified. When these primary batches are not
production scale, a minimum of 6 months of data from production batches should also be submitted to support the evaluation of the regulatory submission. A minimum of 6 months stability data from primary batches should be submitted in cases where shelf life is greater than 6 months. For drug substances and drug products with a shelf life of less than 6 months, the minimum amount of stability data in the initial regulatory submission should be determined on a case-by-case basis. Refer to Section 15 - Stability Considerations for Commitments and Product Lifecyle Management for guidance on providing commitment stability data after marketing authorisation.
生物制品原液和制剂通常需要严格的贮藏条件,以确保维持生物活性并避免降解,这是因为其分子构象和生物活性依赖于共价和非共价力,使其对环境因素(如温度变化、氧化、光照、离子含量和剪切力)极为敏感。对其稳定性的评估可能需要采用复杂的分析方法,包括理化、生化和免疫化学方法,也需要考虑许多可能影响产品效价、纯度和质量的外部条件。对于生物制品原液和制剂,应提交涵盖拟定的有效期的三个注册批次的数据,除非可以解释替代方案的合理性。当这些注册稳定性批次的规模不是生产规模时,还应提交至少6个月的生产批次数据,以支持对注册申报的审评。如果有效期超过6个月,应提交注册稳定性批次至少6个月的稳定性数据。对于有效期小于6个月的原液和制剂,首次注册申报时的稳定性数据最低数量应视具体情况而定。关于在获得上市许可后提供承诺稳定性数据的指导,请参阅第15节“关于承诺和产品生命周期管理的稳定性考虑因素”。
A stability study to establish a re-test period or shelf life should include at least three batches of the drug substance or at least three batches of each strength of the drug product covering the proposed container closure systems. Reduced designs may be applied where justified (refer to Annex 1 – Reduced Stability Protocol Design).
旨在确定复检期/有效期的稳定性研究应包含使用拟定容器密封系统的至少三个原料药/原液批次或每种规格、装量或浓度的至少三个制剂批次。在合理的情况下,可采用简化设计(见附录1-简化稳定性方案设计)。
For synthetic chemical entities and biologicals, if primary batches are not production scale or not all at production scale, the applicant should commit to continuing or initiating and completing a commitment stability study to establish and confirm the re-test period or shelf life in accordance with Section 15.1 - Commitment Stability Studies.
对于化学合成实体和生物制品,如果注册稳定性批次不是生产规模或并非全部处于生产规模,申请人应承诺继续或启动并完成承诺稳定性研究,以根据第15.1节“承诺稳定性研究”建立并确认复检期/有效期。
Table 1: Recommended Core Stability Data for the Standard Approach at Submission to
Support the Initial Re-test Period or Shelf Life1
表1 在申报时用于支持初始复检期/有效期标准方法的建议核心稳定性数据1
|
Product Type |
Batch Type |
Number of Batches2 |
Long-term storage condition |
Accelerated storage condition |
|
New synthetic chemical entity drug substances and/or drug products for which a new drug regulatory submission is required4 |
Primary5 |
3 |
12 months |
6 months3 |
|
Existing synthetic chemical entity drug substances and/or drug products for which an abbreviated/ abridged regulatory submission is required |
Primary5 |
3 |
6 months |
6 months3 |
|
Biological drug substances and/or drug products |
Primary, Production5 |
3 |
6 months 6 |
6 months7 |
|
产品类型 |
批次类型 |
批次数量2 |
长期条件 |
加速条件 |
|
需进行新的药物注册申报的 新化学合成实体原料药和/或制剂4 |
注册稳定性批次5 |
3 |
12个月 |
6个月3 |
|
需进行简化/简略注册申报的 现有化学合成实体原料药和/或相关制剂 |
注册稳定性批次5 |
3 |
6个月 |
6个月3 |
|
生物原液和/或制剂 |
注册稳定性批次,生产批次5 |
3 |
6个月6 |
6个月7 |
1 For testing frequency guidance refer to Section 6 – Testing Frequency
1有关检测频率的指导,请参阅第6节“试验频率”
2 For a full design, at least 3 batches of the drug substance or at least 3 batches of each strength of the drug product covering the proposed container closure systems are tested. Reduced designs may be applied where justified (refer to Annex 1 – Reduced Stability Protocol Design).
2对于完整设计,至少三个原料药/原液批次或至少每种规格或装量生产3个制剂批次,包含建议的容器密封系统。在合理的情况下,可采用简化设计(见附录1-简化稳定性方案设计)
3 If a significant change (refer to Section 13 - Data Evaluation) or an out of specification result occurs at accelerated conditions within the first 3 months, it is considered unnecessary to continue to test through 6 months.
3如果在加速条件下的前3个月内出现显著变化(见第13节“数据评估”)或超标结果,则认为没有必要继续进行为期6个月的试验。
4 In principle, stability protocols for new dosage forms and new strengths/concentrations should follow the guidance for a new drug. However, a reduced stability dataset at submission time (e.g., 6 months accelerated and 6 months long term data) may be acceptable in certain justified cases (refer to Section 15.3 - Stability Studies to Support New Dosage Forms and New Strengths/Concentrations).
4原则上,新剂型和新规格/浓度的稳定性方案应遵循新药的指导。但在某些合理的情况下(见第15.3节“支持新剂型和新规格/浓度的稳定性研究”),申报时简略的稳定性数据集(例如6个月加速试验和6个月长期试验的数据)或许是可以接受的。
5 There should be a commitment to continue stability studies for production batches corresponding to the proposed re-test period or shelf life.
5与拟定复检期/有效期相对应的生产批次,应承诺继续进行稳定性研究。
6 A primary batch can be a production batch but does not need to be a production batch. If the re-test period or shelf life proposed from non-production primary batch data is greater than 6 months, stability data from production batches should be a minimum of 6 months. The shelf life would generally be supported by three primary batches having stability data through to shelf life.
6注册稳定性批次可以是生产批次,但不必须是生产批次。如果非生产注册稳定性批次数据建议的复检期/有效期大于6个月,则生产批次的稳定性数据应至少为6个月。有效期通常由三个具有有效期内稳定性数据的注册稳定性批次支持。
7 Testing under accelerated storage conditions is strongly suggested when appropriate for the storage condition and product type and the minimum time period should be justified by the applicant in accordance with the selected storage conditions. A minimum of three time points, including the initial and final, is recommended.
7当贮藏条件和产品类型适当时,本指导原则强烈建议在加速条件下进行试验,申请人应根据选定的贮藏条件证明最短时间期限的合理性。建议至少设置三个时间点,包括初次时间点和末次时间点。
For drug substances and drug products with intended storage periods of less than the recommendations in Table 1, the minimum amount of stability data in the initial regulatory submission should be determined based on the product-specific risks and in accordance with Section 6 – Testing Frequency.
对于预期的贮藏期比表1中的推荐期限更短的原料药和制剂,在首次注册申报时,应根据产品特定风险,并按照第6节“试验频率”确定最低的稳定性数据量。
3.3 Stability-Indicating Critical Quality Attributes
3.3 稳定性指示关键质量属性
CQAs should be identified using the principles outlined in ICH Q6A, Q6B and ICH Q8-Q11. When designing a stability protocol in support of a drug substance or drug product, information on the CQAs and their target acceptance criteria should already be available. Based on prior knowledge and development data, the applicant should identify the stability-indicating CQAs, which are those attributes that may change upon storage and may impact the functionality and/or quality of the drug substance or drug product.
应根据ICH Q6A, Q6B和ICH Q8-Q11中概述的原则鉴别CQA。在设计支持原料药或制剂的稳定性方案时,应已获得关于CQA及其目标可接受标准的信息。根据先验知识和开发数据,申请人应确定指示稳定性的CQA,这些属性可能在贮藏后发生变化,并可能影响原料药或制剂的功能性和/或质量。
3.3.1 Recommendations for Establishing a Re-Test Period or Shelf life.
3.3.1 关于确定复检期/有效期的建议
The stability protocol to establish a re-test period or shelf life should include stability-indicating CQAs and compile a suitable dataset to demonstrate product quality through storage and use. For synthetic chemical drug substances and drug products, the stability protocol should consider appropriate, physical and chemical attributes. For biological drug substances and drug products, the protocol should assess changes in CQAs that affect physicochemical properties, purity and impurity levels, immunochemical properties and the biological activity of the product, as appropriate. For both synthetics and biologicals, microbiological attributes and product performance characteristics should be confirmed on stability as applicable. For products that are particularly sensitive to changes in temperature, oxidation, light, moisture content and shear forces, quality attributes that may be impacted should be assessed. For additional information on attributes to be included in the drug substance or drug product specification, refer to ICH Q6A and Q6B.
用于确定复检期/有效期的稳定性方案应包括指示稳定性的CQA,并汇编适当的数据集以证明在贮藏和使用过程中的产品质量。对于合成化学原料药和制剂,稳定性方案应酌情考虑理化属性。对于生物制品的原液和制剂,方案应酌情评估影响产品理化性质、纯度和杂质水平、免疫化学性质和生物活性的CQA的变化。对于化学合成实体和生物制品,应酌情确认稳定性的微生物属性和产品性能特征。对于对温度、氧化、光照、水分含量和剪切力变化特别敏感的产品,应评估其可能受影响的质量属性。有关原料药或制剂质量标准中包含的质量属性的其他信息,请参考ICH Q6A和Q6B。
Where excipient levels or their properties may change on stability, potentially impacting drug product CQAs, they should be evaluated as part of drug product stability testing, (e.g., levels of surfactant, preservative content). In cases where stabilisers are needed for a biological drug substance, the same considerations should be applied. Co-packaged diluents should follow the recommendations for drug products. A risk-based approach is recommended, where development data and excipient prior knowledge can be used to understand whether additional drug substance and/or drug product stability data are appropriate to support the re-test period or shelf life.
如果辅料水平或其特性在稳定性期间可能发生变化,从而可能影响制剂CQA,则应将其作为制剂稳定性试验的一部分进行评估(比如表面活性剂的水平和防腐剂含量)。对于生物制品需要使用稳定剂的情况,也应进行同样的考量。合并包装的稀释剂将遵循针对制剂的建议。建议采用基于风险的方法,其中开发数据和辅料先验知识可用于了解是否需要额外的原料药和/或制剂稳定性数据, 合理支持复检期或有效期。
In accordance with the principles outlined in ICH Q3D and Q3E, stability-indicating CQAs
considerations should include potential interaction with the respective storage container, contact with administration or delivery devices (e.g., syringe walls, catheters and injection needle) and dispersion media (such as solvents for reconstitution or dilution).
根据ICH Q3D和Q3E中概述的原则,指示稳定性的CQAs考虑因素应包括与相应贮藏容器的潜在相互作用、与给药或递送装置(如注射器壁、导管和注射针)和分散介质(如复溶溶剂或稀释剂)之间的接触。
3.3.2 Recommendation for Lifecycle Stability Protocols
3.3.2 生命周期稳定性方案建议
After additional knowledge is gained following establishment of the re-test period or shelf life, data may confirm that some CQAs do not change on stability and stability protocols to support the product lifecycle may be updated accordingly (refer to Section 15 – Stability Considerations for Commitments and Product Lifecyle and Annex 1 - Reduced Stability Protocol Design)
在确定复检期/有效期并获得更多信息后,数据可确认某些CQAs是否在稳定性研究中未发生变化,并相应更新支持产品生命周期的稳定性方案(见第15节“关于承诺和产品生命周期管理的稳定性考虑因素”
3.4 Specifications
3.4 质量标准
3.4.1 Tests and Analytical Procedures
3.4.1 测试和分析程序
Before a formal stability study protocol is initiated, the suitability of the proposed analytical procedures to detect changes in the stability-indicating CQAs should be assessed in accordance with ICH Q2 and ICH Q14. The analytical procedures used to monitor changes in the stability-indicating CQAs should be chosen and validated to provide assurance that changes to product quality will be detected, measured and understood over the expected re-test period or shelf life. Establishment of potential degradation pathways (refer to Section 2.3 -Analysis and Interpretation of Results) is important when developing and validating suitable analytical procedures. When feasible for synthetic chemical entities, the mass
balance relationship between tested attributes should be observed when selecting appropriate stability- indicating tests. For example, for solid drug substances or drug products, an apparent decrease in the active moiety could be caused by an increase in degradation products and/or an increase in moisture content.
在启动正式稳定性研究的方案之前,应根据ICH Q2和ICH Q14评估用建议的分析方法检测指示稳定性的CQA变化的适用性。应选择并验证用于监测指示稳定性的CQA变化的分析方法,以确保在预期的复检期/有效期内检测、测量和理解产品质量的变化。在开发和验证合适的分析方法时,确定潜在的降解途径(见第2.3节“结果分析与解读”)非常重要。如果对于化学合成实体可行,在选择适当的指示稳定性试验时,应观察被测属性之间的质量平衡关系。例如,对于固体原料药或制剂,活性部分的明显减少可能是由降解产物的增加和/或水分含量的增加引起的。
When justified, the analytical procedures used for stability testing may differ from the release analytical procedure for the same quality attribute (e.g., container closure integrity testing may be used instead of sterility testing during stability). In situations where stability-indicating quality attributes are not tested as part of release testing (e.g., the relevant CQAs are measured and controlled during processing as described in ICH Q8), additional analytical procedures should be established to support stability studies.
在合理的情况下,用于稳定性测试的分析程序可与相同质量属性的放行分析程序不同(比如在稳定性研究期间可能使用容器密封完整性测试代替无菌测试)。在未将指示稳定性的质量属性作为放行检测的一部分进行检测的情况下(比如按照ICH Q8所述在处理过程中测量和控制相关CQA),应建立额外的分析方法来支持稳定性研究。
3.4.2 Acceptance Criteria
3.4.2 可接受标准
The shelf life acceptance criteria should consider all available stability information from development and manufacture of the drug substance through final drug product shelf life in accordance with ICH Q6A and Q6B. As per these guidelines, when a stability-indicating CQA changes over time, it may be appropriate to establish a release specification that is more stringent than the shelf life specification to ensure that the drug substance and/or drug product quality is maintained through to the end of shelf life.
In general, any differences between the release and shelf life acceptance criteria should be justified with data. In case a re-test period is assigned to a drug substance, generally the acceptance criteria are the same as at release.
根据ICH Q6A和Q6B,有效期可接受标准的制定应考虑从原料药/原液开发和生产到最终制剂有效期的所有可用稳定性信息。根据这些指导原则,当指示稳定性的关键质量属性随时间变化时,可能需要建立比有效期质量标准更为严格的放行标准,以确保原料药和/或制剂能够在有效期结束前一直维持良好质量。一般来说,放行标准和有效期可接受标准之间的任何差异都应该用数据来证明。如果为原料药指定了复检期,则其可接受标准通常与放行标准相同。
3.4.3 Pharmacopoeial Critical Quality Attributes and Analytical Procedures
3.4.3 药典关键质量属性和分析方法
When drug substance and/or drug product monographs or general procedures are available and relevant to the region(s) where the regulatory submission is to be filed, the monographed CQAs and analytical procedures are an appropriate starting point in designing a product-specific stability protocol. Any differences in the proposed analytical procedures from those in the pharmacopeia should be scientifically justified (e.g., including demonstration of equivalency). A knowledge- and risk-based approach should then be applied to ensure that any differences in stability behaviour are properly controlled.
当原料药/原液和/或制剂专论或常规方法在拟提交注册申报的地区可用并相关时,专论CQA和分析方法可以作为设计产品特定稳定性方案的合适起点。建议的分析方法与药典方法的任何差异都应有科学依据(比如包括等效性证明)。应该采用基于知识和风险的策略,以确保任何稳定性行动之间的差异都能得到有效控制。
3.5 Additional Considerations for Vaccines
3.5 关于疫苗的其他考虑因素
In cases where the potency of the product is dependent on conjugation and/or adsorption of the active ingredient to another moiety (e.g., carrier), applicants should evaluate potential dissociation of the active ingredient(s) from the carrier during storage (e.g., in conjugate vaccines).
如果制剂的效价取决于活性成分与另一个部分(如载体)的结合和/或吸附,申请人应评估在贮藏期间活性成分与载体可能的解离情况(比如在结合疫苗中解离)。
In cases where the potency of the product is dependent on the inclusion of an adjuvant, the CQAs for the adjuvant should be evaluated during stability studies.
如果产品的效价取决于是否含有佐剂,则应在稳定性研究期间评估佐剂的CQA。
It is strongly recommended that stability studies for vaccines include mechanisms to evaluate the potency (i.e., the specific ability or capacity to achieve its intended effect using suitable methods) of the product.
本指导原则强烈建议在关于疫苗的稳定性研究中纳入评估产品效价的机制(即用适当方法达到预期效果的特定能力或容量)。
3.6 Additional Considerations for the Combination of a Drug Product with a Medical Device
3.6 药械组合的其他考虑因素
The stability of a combination of a drug product with a medical device considers (a) drug product CQAs and (b) drug device combination performance characteristics through storage to the completion of administration (refer to Section 11 – In Use Stability). The functional performance characteristics of the device constituent alone are outside of the scope of this guideline and are addressed through device design verification studies.
药械组合的稳定性考虑因素包括:(a)制剂CQA和(b)从贮藏至给药完成期间的药械组合性能特征(见第11节“使用中稳定性”)。设备组件单独的功能性能特征不在本指导原则的范围内,需通过组件设计验证研究加以解决。
The stability protocol design for a combination of a drug product with a medical device (integral or co-packaged) should follow the same principles as described for a drug product, including a risk assessment and compatibility with contact materials. Stability-indicating attributes of the drug constituent may impact the medical device functional performance characteristics, and stability studies and conclusions should account for these interactions. Considerations should be made for the administration-dependent functional performance characteristics of the fully assembled combination of a drug product with a medical device that may be impacted by long-term storage (i.e., CQAs that can only be assessed after
assembly). The storage orientation may be established based on a risk assessment. The shelf life of a co-packaged combination of a drug product with a medical device should be based on the shorter of either the device constituent part or the drug constituent part shelf life. For integrated device-drug products, the shelf life should be based on the shorter of either of the constituent part or the final combination of a drug product with a medical device.
药械组合(单一实体或组合包装)的稳定性方案设计应遵循与制剂相同的原则,包括风险评估以及与直接接触材料的相容性。药物成分的稳定性指示属性可能会影响医疗器械功能性能特征,稳定性研究和结论应对这些相互作用做出解释。应该考虑制剂与医疗器械完全组装后的组合在给药依赖下的功能性能特征,这些特征可能会受到长期贮藏的影响(即只能在组装后评估的关键质量属性)。可根据风险评估确定贮藏考量。组合包装的药械组合的有效期应以器械组成部分或制剂有效期中的较短者为准。对于一体式包装的药械组合,有效期应以器械组成部分或最终药械组合制剂有效期中的较短者为准。
制剂与医疗器械的每种组合类型都有其独特的质量属性和基于给药方式的功能性能特征列表。
Each type of combination of a drug product with a medical device should have its own unique list of quality attributes and administration-dependent functional performance characteristics. Attributes should be risk assessed according to the specific design of that product to identify the critical attributes or characteristics. The risk assessment may include data from device design development studies and prior knowledge from similar combinations of a drug product with a medical device. The stability protocol should use the assembled (integral or co-packaged) product representative of the product proposed for marketing. If the stability studies were not performed with the combination of a drug product with a medical device as proposed for marketing, the changes made should be assessed and justified with respect to the impact on stability.
应根据产品的具体设计对其属性进行风险评估,以确定关键属性或特性。风险评估可能包括器械设计开发研究的数据以及制剂与医疗器械类似组合的先验知识。稳定性方案应使用代表拟上市产品的组装(一体式包装或组合包装)产品。如果未以拟上市组合形式进行稳定性研究,则应对相应变更进行评估,并说明其对稳定性的影响。
3.7 Risk Management
3.7 风险管理
A science- and risk-based approach should be used to inform the different aspects of protocol design outlined in Section 4 - Selection of Batches through Section 7 - Storage Conditions.
应采用基于科学和风险的方法,为第4节“批次选择”至第7节“贮藏条件”中概述的方案设计的提供不同的决策依据。
The inclusion of risk management information with a registered stability protocol is not mandatory, but in cases where it forms the basis of a justification for enhanced/reduced protocol approaches, information on the risk assessment process, outcome and the connection to the stability protocol should be described.
尽管不强制要求在注册稳定性方案中纳入风险管理信息,但如果该信息构成扩充或简略方案方法的依据,则应描述风险评估过程、风险评估结果及其与稳定性方案的关联信息。
4 SELECTION OF BATCHES
4 批次选择
To establish a re-test period or shelf life for the drug substance and drug product, stability data should generally be provided on three primary batches. Alternative approaches for batch requirements may be supported when justified. The manufacturing process for the primary batches of drug substance and drug product should be similar or representative, but not necessarily identical to the manufacturing process used for production batches. Hence, a primary batch may be but is not necessarily a production batch. Differences in the manufacturing processes for the primary batches and those proposed for production batches should be justified. Specific considerations for primary stability batches are provided in Table 2.
为建立原料药和制剂的复检期或有效期,通常应提供三个注册稳定性批次的稳定性数据。在合理的情况下,可支持批量要求的替代方案。原料药和制剂注册稳定性批次的生产工艺应当相似或具有代表性,但不一定与用于生产批次的生产工艺完全相同。因此,注册稳定性批次可以是生产批次,但不一定是生产批次。注册稳定性批次和拟生产批次的生产工艺差异应合理。注册稳定性批次的具体考虑因素见表2。
For studies that are not a primary study (e.g., in-use stability, photostability, supportive studies and stability studies to support post-approval changes) and use non-production batches, the batches should be representative as described below:
对于非主要研究(例如,使用中稳定性、光稳定性、支持性研究和支持批准后变更的稳定性研究)和使用非生产批次的研究,使用的批次应具有代表性,如下所述:
• Synthetic chemical entities: Chemically synthesised drug substances should be manufactured by the same synthetic route. Changes to manufacturing process parameters should be scientifically justified. Drug products should be of the same formulation and method of manufacture.
• 化学合成实体:化学合成的原料药应采用相同的合成路线进行生产。生产工艺参数
的变更应有科学依据。制剂的处方和生产方法应相同。
• Biologicals: The quality of all drug substance and drug product batches placed in a stability program should be manufactured using a similar process to the proposed production manufacturing process and be analytically comparable to the production batches (refer to ICH Q5E). The analytical comparability for the clinical batches and the non-production batches to the production batches should be demonstrated. A comprehensive analytical comparability exercise may include additional characterisation testing.
• 生物制品:稳定性计划中的所有原液和制剂批次应采用与建议生产工艺相似的工艺进行生产,并在分析上与生产批次具有可比性(见ICH Q5E)。应证明临床批次和非生产批次与生产批次的分析可比性。全面的分析可比性研究可能包括额外的特性鉴定试验。
4.1 Considerations for Selection of Primary Stability Batches
4.1 注册稳定性批次选择的考虑因素
Where possible, batches of drug product included in stability testing should be derived from different batches of drug substance to account for variability in drug substance batches. Stability studies should be performed on each individual strength, fill volume and container closure system of the drug product unless a reduced protocol design is applied (refer to Annex 1 – Reduced Stability Protocol Design).
在可能的情况下,稳定性试验中包含的制剂批次应来自不同的原料药批次,以将原料药批次的变异性纳入考虑。应对制剂的每种规格、装量和容器密封系统进行稳定性研究,除非采用了简略方案设计(见附录1-简化稳定性方案设计)。
The primary stability batches of the drug substance and drug product should be representative of the clinical and production batches as described above. Additional development batches that are representative of the primary and production batches may also be included as supporting stability data.
原料药/原液和制剂的注册稳定性批次应代表上述临床和生产批次。代表申报批次和生产批次的其他开发批次也可作为支持性稳定性数据。
Refer to Table 2 below for additional considerations at time of selection of primary stability batches.
选择注册稳定性批次时的其他考虑因素见下表2。
Table 2: Considerations for Primary Stability Batches of Drug Substance and Drug Product
表2 原料药和制剂注册稳定性批次的考虑因素
|
|
Synthetic Chemical Entities |
Biologicals |
|
Drug Substance |
Same chemical synthetic route Similar manufacturing process (differences justified) At minimum, all batches manufactured at pilot scale2 Meet proposed registration specification Containers constructed of the same material and type of container closure system as production batches. |
Same cell production system, if applicable Similar manufacturing process (differences justified) Meet proposed registration release specification Containers constructed of the same material and type of container closure system as production batches. Comparable to production batches (ICH Q5E) |
|
Drug Product |
Same formulation1 and dosage form Minimum of 2 batches manufactured to at least pilot scale2, other batch(es) can be smaller if justified Same manufacturing process with equipment with the same operating principles. Meet the proposed registration release specification Same fill unless a reduced protocol design is applied1 Same container closure system as proposed for marketing |
Same formulation and dosage form Comparable to production batches (e.g., ICH Q5E) Meet proposed registration release specification Same fill volume unless a reduced protocol design is applied1 Same container closure system as proposed for marketing. |
|
|
化学合成实体 |
生物制剂 |
|
原料药 |
相同的化学合成路线 相似的生产工艺(差异合理) 至少,所有批次均以中试规模生产2 符合拟定的注册标准 由与生产批次采用相同材料和类型的容器密封系统构成的容器。 |
相同的细胞生产系统(如适用) 相似的生产工艺(差异合理) 符合拟定的注册放行标准 由与生产批次采用相同材料和类型的容器密封系统构成的容器。 与生产批次具有可比性(ICH Q5E) |
|
制剂 |
处方1和剂型相同 至少生产2个中试规模批次2,如果有合理依据,可以减小其他批次规模 生产工艺相同,设备操作原理相同。 符合拟定的注册放行标准 除非采用简化的方案设计,否则装量相同1 与拟上市的容器密封系统相同 |
处方和剂型相同 与生产批次具有可比性(比如ICH Q5E) 符合拟定的注册放行标准 除非采用简化的方案设计,否则设计装量相同1 与拟上市的容器密封系统相同。 |
1Refer to Annex 1 – Reduced Stability Protocol Design for details around when exceptions may apply
1有关可能适用例外情况的详细信息,请参阅附录1-简化稳定性方案设计
2In accordance with ICH Q13, the definition of a pilot batch for synthetics does not apply for continuous manufacturing.
2根据ICH Q13,化学合成实体中试批次的定义不适用于连续生产。
When the long-term stability data do not cover the proposed re-test period or shelf life at the time the marketing application is submitted, refer to Section 15 - Stability Considerations for Commitments and Product Lifecycle Management for relevant commitments.
在提交上市申请时,长期稳定性数据未包含建议的复检期/有效期,相关承诺见第15节“关于承诺和产品生命周期管理的稳定性考虑因素”。
4.2 Considerations for Multiple Production Sites in the Initial Regulatory Submission
4.2 首次注册申报中对多个生产场地的考虑因素
The stability data from each site, provided in the initial regulatory submission should be proportionate to the overall product, process and facility risk and in accordance with regional requirements. For both synthetics and biologicals, when the product, process and production site are comparable, the re-test period and/or shelf life would not need to be re-established at an additional production site. An additional production site refers to any production site proposed in the initial regulatory submission other than the drug substance and drug product site where the original production scale batches are manufactured.
首次注册申报中提供的每个场地的稳定性数据应与整体的产品、工艺和设施风险成比例,并符合地区要求。对于化学合成实体和生物制品,当产品、工艺和生产场地具有可比性时,不需要在其他生产场地重新确定复检期和/或有效期。额外生产场地是指除用于生产初始生产规模批次的原料药和制剂场地外的,首次注册申报时建议的任何生产场地。
For synthetic chemical entities, a comparison of batch data of the primary batches with data from each production site should be provided in the regulatory submission. The amount of stability data provided for each production site depends on the risk associated with implementing each additional production site for the drug substance or drug product. A commitment stability study should be established for each production site in accordance with Section 15.1 - Commitment Stability Studies. The number of production batches from each site in the commitment stability study can be fewer than three with a supporting scientific justification and risk assessment.
对于化学合成实体,应在注册申报中提供注册稳定性批次的批次数据与每个生产场地数据的比较情况。为每个生产场地提供的稳定性数据批次数量取决于与实施原料药或制剂的每个额外生产场地相关的风险。应根据第15.1节“承诺稳定性研究”,为每个生产场地建立承诺稳定性研究。在承诺稳定性研究中,每个生产场地的生产批次数量可以少于三个,但需要具有支持性的科学依据和风险评估。
For biologicals, the default minimum stability data presented for each production site, should be as outlined in Table 1 of Section 3.2 -Recommended Minimum Core Stability Data for the Standard Approach at Submission to Support Initial Re-test Period or Shelf Life. However, for biologicals with an enhanced level of product and process understanding, an alternative science- and risk-based approach may be justified for those additional sites that are receiving the transferred manufacturing process from an originating production site. A comparability assessment inclusive of accelerated and/or stressed condition stability results for commercial scale production batches manufactured at the proposed additional site relative to primary batches from the original production site should be provided (refer to ICH Q5E). Based on risk assessment that considers analytical comparability, process comparability and production site history for the manufacture of similar product types, sites receiving the transferred manufacturing process may initially propose a reduced number of production scale stability studies in the regulatory submission. When a reduced data set is justified, a commitment should be made to continue stability studies at each site through the proposed re-test period or shelf life for a total of three production scale batches in accordance with Section 15.1 - Commitment Stability Studies.
对于生物制品,每个生产场地的默认最低稳定性数据应如第3.2节中的表1“在申报时用于支持初始复检期/有效期标准方法的建议最低核心稳定性数据”所述一致。然而,对于产品和工艺已得到深入理解的生物制品而言,其他的、从初始生产场地接收和转移制造工艺的生产场地采用科学的、基于风险的替代方案却可能是更合理的。应提供可比性评估,包括在建议的额外场地生产的商业规模生产批次相对于初始生产场地的注册稳定性批次在加速和/或强力条件下的稳定性结果(见ICH Q5E)。根据风险评估,并考虑到分析的可比性、工艺的可比性和生产类似产品类型的生产场地历史,接收生产工艺转移的场地起初可在注册申报中建议减少生产规模稳定性研究的数量。如果简化的数据集是合理的,则应承诺按照第15.1节“承诺稳定性研究”,继续在每个生产场地进行总共三个生产规模批次的稳定性研究至拟定复检期/货架期。
4.3 Considerations for Vaccines
4.3 疫苗的考虑因素
In general, production scale batches are expected to be used to set shelf life of vaccines. If non- production scale batches are used as primary batches, a justification should be based on product knowledge, comparability studies and risk. The remaining recommendations for primary batches for biologicals in Table 2 are also applicable to vaccines.
一般来说,使用生产规模批次来设定疫苗的有效期。如果使用非生产规模批次作为注册稳定性批次,则应根据产品知识、可比性研究和风险进行论证。表2中关于生物制品注册稳定性批次的其余建议也适用于疫苗。
4.4 Considerations for Continuous Manufacturing Processes
4.4 连续制造工艺的考虑因素
For guidance on selection of batches from a CM process, refer to ICH Q13 guideline. For recombinant protein biologicals, the use of a single start-up/shutdown sequence (refer to ICH Q13) to manufacture multiple primary drug substance stability batches is typically not applicable. Primary drug substance stability batches should be obtained from multiple harvests/cell bank thaws and should cover the entire cell culture duration. The drug product primary stability batches manufactured by CM processes should incorporate the variability described for different drug substance batches.
关于从连续制造(CM)工艺中批次选择的指导,请参考ICH Q13指导原则。对于重组蛋白生物制品,使用单一启动/关闭顺序(见ICH Q13)来生产多个原液注册稳定性批次的方法通常不适用。原液注册稳定性批次应从多次收获/细胞库解冻中获得,并应涵盖整个细胞培养过程所用时间。通过CM工艺生产的制剂注册稳定性批次应包括不同原液批次的变异性。
5 CONTAINER CLOSURE SYSTEM
5 容器密封系统
A container closure system comprises the primary (in contact with the product) and the secondary packaging if the latter are functional (e.g., combination of a drug product with a medical device) or intended to provide additional protection to the drug product. The stability study design should consider and include the secondary package when it is protective or directly impacts the chemical, physical, or functional attributes, unless otherwise justified.
容器密封系统包括内包装(与产品直接接触)和次级包装,前提是后者具有一定功能(比如药械组合),或旨在为药品提供额外的保护。除非另有说明,否则当次级包装具有保护性或直接影响产品的化学、物理或功能属性时,稳定性研究设计应考虑并纳入次级包装。
The primary stability studies for the drug substance should be conducted in a container closure system that is the same or representative of the packaging proposed for storage and distribution. The container closure system should be the same type and constructed of the same material as production batches (dimensions may be smaller). For the drug product, the commercial container closure is recommended to ensure that the proposed container closure system can adequately protect the dosage form, is compatible with the dosage form and will function in the manner for which it is designed through a product's intended shelf life. When applicable, impact of packaging components from which matter may migrate into the product (e.g., ink or adhesive from labels) should also be considered.
原料药的注册稳定性研究应在与拟用于贮藏和分销的包装相同或具有代表性的容器密封系统中进行。容器密封系统的类型和材质应与生产批次相同(尺寸可能更小)。对于制剂,建议采用商业容器密封系统,以确保建议的容器密封系统能够充分保护剂型、与剂型相容,并在产品预期有效期内按照设计的方式发挥作用。在适用的情况下,还应考虑可能迁移到产品中的物质的包装组件的影响(比如标签上的墨水或粘合剂)。
Changes in the quality of a product may occur due to the interactions between the drug substance or drug product and the respective container closure system, and the effect of such interactions on product stability should be evaluated. Any impact of container orientation on the critical quality attributes of the drug product should be assessed based on prior knowledge gained through development and/or as part of stability studies. For primary batches of liquids, solutions, semi-solids and suspensions, the product should be placed into an inverted (or horizontal) position and an upright (or vertical) position unless a worst-case orientation is justified with supporting data. However, when drug product-container closure
interactions cannot be excluded, stability studies should include samples maintained in both the inverted (or horizontal) position, as well as in the upright (or vertical) position (e.g., when storage orientation can have a significant effect on the delivered dose/repriming period of pressurised metered dose inhalers).
原料药或制剂同相应的容器密封系统之间的相互作用可能会导致产品质量发生变化,应评估这种相互作用对产品稳定性的影响。应根据通过开发获得的先验知识和/或作为稳定性研究的一部分,评估容器放置方向对制剂关键质量属性有何影响。对于液体、溶液、半固体和悬浮液的注册稳定性批次,产品应以倒置(或水平)放置和直立(或垂直)方式放置,除非有支持性数据证明提出的最差情况下的方向是合理的。然而,当无法排除制剂-容器密封系统相互作用时,稳定性研究应包括样品在倒置(或水平)位置和直立(或垂直)位置的贮藏(比如当贮藏方向对加压定量吸入器的剂量/再启动期有显著影响时)。
6 TESTING FREQUENCY
6 检测频率
The proposed protocol should align with the principles outlined in Section 13 - Data Evaluation and include sufficient timepoints to verify any proposed extrapolation or stability model, where appropriate for the product type.
建议方案应符合第13节“数据评价”中概述的原则,并包括足够的时间点,以验证适用于产品类型的任何建议外推或稳定性模型。
For primary stability studies, the frequency of testing should be sufficient to establish the stability profile of the drug substance or drug product. For a drug substance or drug product with a proposed re-test period/shelf life of 12 months or less, the frequency of testing at the long-term storage condition is recommended monthly for the first 3 months and at 3-month intervals thereafter. For cases when an intended re-test period/shelf life is very short, sufficient time points should be considered. For a drug substance or drug product with a proposed re-test period/shelf life greater than 12 months, the recommended frequency of testing at the long-term storage condition should normally be every 3 months over the first year, every 6 months over the second year and annually thereafter through to the end of the proposed re-test period/shelf life. Sterility testing or alternatives (e.g., container closure integrity testing) should be performed at a minimum annually, including initially and at the end of the proposed re-test period or shelf life.
对于注册稳定性研究,检测频率应足以建立原料药或制剂的稳定性特征。对于建议复检期/有效期为12个月或更短的原料药或制剂,在长期条件下建议前3个月每月检测一次,以后每隔3个月检测一次。对于预期的复检期/有效期较短的情况,应考虑足够的时间点。对于建议复检期/有效期大于12个月的原料药或制剂,在长期条件下的建议试验检测频率通常为第一年每3个月一次,第二年每6个月一次,此后每年一次,直至建议复检期/有效期结束。无菌测试或替代测试(如容器密封完整性测试)应至少每年进行一次,包括初始和建议的复检期/有效期结束时。
For studies under accelerated conditions, a minimum of three time points, including the initial and final time points, is recommended (e.g., 0, 3 and 6 months is recommended for a 6-month study). Where an expectation (e.g., based on development experience) exists that results from accelerated studies are likely to approach significant change criteria (refer to Section 13 – Data Evaluation) or likely to be out of specification, increased testing is recommended. Increased testing could be conducted either by (a) including an additional less severe temperature condition (i.e., intermediate) that may be better predictive of the long-term stability and /or (b) including an additional time point in the accelerated study design which may be earlier than the final time point. Note that this would not preclude following the recommendations in Section 13 - Data Evaluation, when deciding whether extrapolation is
applicable. At the intermediate storage condition, a minimum of four time points, including the initial and final time points (e.g., 0, 6, 9 and 12 months, from a 12-month study) is recommended.
对于加速条件下的研究,建议至少设置三个时间点,包括初次时间点和末次时间点(比如对一项考察期为6个月的研究而言,建议将时间点设置为0、3和6个月)。如果预计(比如基于开发经验)加速研究的结果可能接近显著变化标准(见第13节“数据评估”)或可能超出质量标准,则建议增加试验。增加的试验可通过以下方式进行:(a)纳入一个额外的不太严格的温度条件(即中间温度),这可能更好地预测长期稳定性;和/或(b)在加速研究设计中纳入一个额外的时间点,该时间点可能早于末次时间点。请注意,在决定外推是否适用时,并不排除要遵循第13节“数据评估”中的建议。在中间条件下,建议至少设置四个时间点,包括初次和末次时间点(比如对一项考察期为12个月的研究而言,建议将时间点设置为0、6、9和12个月)。
As discussed in Annex 1 - Reduced Stability Protocol Design and Section 15.3 - Product Lifecycle Stability Studies, a reduced testing frequency may be justified when potential stability-indicating CQAs show no change over time. The minimum testing frequency recommended in this section may not be applicable if alternative strategies are applied (refer to Section 13 – Data Evaluation and Annex 2 –Stability Modelling).
如附录1-简化稳定性方案设计和第15.3节“产品生命周期稳定性研究”中所述,当潜在指示稳定性的CQA未随时间变化时,可证明降低试验频率是合理的。如果采用替代策略,本节中推荐的最低试验频率可能不适用(见第13节“数据评估”和附录2-稳定性建模)。
7 STORAGE CONDITIONS
7 贮藏条件
7.1 General Considerations
7.1 一般考虑因素
Stability of drug substances and drug products should be evaluated under storage conditions with appropriate tolerances that test for thermal and moisture stability and, if applicable, sensitivity to potential solvent loss. For sensitivity to light, refer to Section 8 – Photostability. The storage conditions and the duration of studies chosen should cover the intended storage and use, including considerations for shipment and any short-term storage condition (refer to Section 10 – Short-Term Storage Conditions). Advice on storage conditions to support an in-use period is detailed in Section 11 - In-Use Stability.
应在具有适当耐受性的贮藏条件下对原料药和制剂的稳定性进行评估,以检测其热稳定性和湿度稳定性,以及对潜在溶剂损失的敏感性(如适用)。关于对光的敏感性,请参阅第8节“光稳定性”。所选择的贮藏条件和研究持续时间应涵盖预期的贮藏和情况,包括对运输以及任何短期贮存条件的考虑(见第10节“短期条件”)。第11节“使用中稳定性”中详细介绍了支持使用中保存条件建议。
Testing at accelerated conditions or stress testing is essential to establish product stability information, such as to establish the degradation pathways and the intrinsic stability of the molecule, to confirm the stability-indicating nature of the analytical procedures (refer to Section 2 – Development Studies Under Stress and Forced Conditions and Section 3.3 – Stability-Indicating Critical Quality Attributes) and unintended excursions in storage conditions. Data generated under accelerated conditions may enable stability modelling. Accelerated conditions data may support extrapolation of the intended re-test periodand shelf life (refer to Section 13 – Data Evaluation).
加速条件下的试验或强制降解试验对于确定产品稳定性信息至关重要,例如确定降解途径、和分子的固有稳定性、确认分析方法的稳定性指示性质(见第2节“强力条件和强制降解条件下的开发研究”和第3.3节“指示稳定性的关键质量属性”)和贮藏条件下的非预期偏离。在加速条件下生成的数据可以实现稳定性建模。加速条件数据可支持对预期的复检期和有效期进行外推(见第13节“数据评估”)。
Since most biologicals are sensitive to physical conditions, data obtained under accelerated conditions may confirm the stability-indicating nature of the analytical procedures or help elucidate the degradation profile of a biological drug substance or drug product. Data from accelerated conditions could also support that a manufacturing change did not impact the stability profile.
由于大多数生物制品都对贮藏条件敏感,因此在加速条件下获得的数据可证实分析方法的稳定性指示性质,或有助于了解生物原料药或制剂的降解产物谱。加速条件下的数据也可为生产变更没有影响稳定性特征提供支持。
Where it can be justified that a proposed container closure system and conditions of storage afford sufficient protection against high and low humidity conditions, stability studies at different relative humidities can usually be omitted. Appropriate stability data under recommended storage conditions should be provided if containers other than impermeable containers are used.
如果能够证明建议的容器密封系统和贮藏条件在高湿度和低湿度条件下提供了足够的保护,则通常可以省略不同相对湿度下的稳定性研究。如果使用非渗透性容器以外的容器,应提供推荐贮藏条件下的适当稳定性数据。
The storage conditions to be applied to the different stability studies are detailed in the sections below.The equipment utilised should be capable of controlling the storage condition within the ranges defined in this guideline. The actual temperature and humidity (when controlled) should be monitored during stability storage. Short-term spikes due to opening of doors of the storage facility are accepted as unavoidable. The effect of excursions due to equipment failure should be addressed and reported if judged to affect stability results. Excursions that exceed the defined tolerances for more than 24 hours should be described in the study report and their effect assessed.
适用于不同稳定性研究的贮藏条件详见以下章节。所使用的设备应能将贮藏条件控制在本指导原则规定的范围内。在稳定性贮藏期间,应在实际温度和湿度(得到控制时)对其进行监测。由于贮藏设施的门打开而导致出现的短期峰值被认为是不可避免的。如果判断设备故障导致的偏移影响到稳定性结果,则应说明并报告该偏移的影响。应在研究报告中描述超过规定24小时的偏移,并评估其影响。
Alternative storage conditions can be used if justified. Recommendations are applicable to both synthetic chemical entities and biological products, unless otherwise specified.
如果合理,可以使用其他贮藏条件。除非另有规定,否则建议适用于化学合成实体和生物制品。
7.2 Considerations for Products Intended to be Stored at Room Temperature
7.2 拟在室温下贮藏产品的注意事项
The recommended storage conditions that are applicable to each climatic zone are outlined in the table below.
下表列出了适用于每个气候带的推荐贮藏条件。
Table 3: Storage Condition Recommendations for Each Climatic Zone1
表 3各气候带的贮藏条件建议
|
Climatic Zone1 |
Long-term2 |
Intermediate |
Accelerated |
|
I and II |
25°C ±2°C/60% RH ±5% RH |
30°C ±2°C/65% RH ±5% RH, or 30°C ±2°C/75% RH ±5% RH |
40°C ±2°C/75% RH ±5% RH |
|
30°C ±2°C/65% RH ±5% RH, or 30°C ±2°C/75% RH ±5% RH |
Not applicable |
40°C ±2°C/75% RH ±5% RH |
|
|
III |
30°C ±2°C/35% RH ±5% RH, or 30°C ±2°C/65% RH ±5% RH, or 30°C ±2°C/75% RH ±5% RH |
Not applicable |
40°C ±2°C/75% RH ±5% RH |
|
IVa |
30°C ±2°C/65% RH ±5% RH, or 30°C ±2°C/75% RH ±5% RH |
Not applicable |
40°C ±2°C/75% RH ±5% RH |
|
IVb |
30°C ±2°C/75% RH ±5% RH |
Not applicable |
40°C ±2°C/75% RH ±5% RH |
|
气候带1 |
长期2 |
中期 |
加速 |
|
I和II |
25°C ±2°C/60% RH ±5% RH |
30°C ±2°C/65% RH ±5% RH, 或 30°C ±2°C/75% RH ±5% RH |
40°C ±2°C/75% RH ±5% RH |
|
30°C ±2°C/65% RH ±5% RH, 或 30°C ±2°C/75% RH ±5% RH |
不适用 |
40°C ±2°C/75% RH ±5% RH |
|
|
III |
30°C ±2°C/35% RH ±5% RH, 或 30°C ±2°C/65% RH ±5% RH, 或 30°C ±2°C/75% RH ±5% RH |
不适用 |
40°C ±2°C/75% RH ±5% RH |
|
IVa |
30°C ±2°C/65% RH ±5% RH, 或 30°C ±2°C/75% RH ±5% RH |
不适用 |
40°C ±2°C/75% RH ±5% RH |
|
IVb |
30°C ±2°C/75% RH ±5% RH |
不适用 |
40°C ±2°C/75% RH ±5% RH |
1Specific regional requirements for more severe storage conditions may however apply
1然而,对于更苛刻贮藏条件,可能适用特定区域要求
2Refer to Section 1.3 – Introduction to Guideline and General Principles
2参见第1.3节-指导原则和一般原则简介
The applicant should determine and justify the long-term stability studies conditions to be performed.
申请人应确定并证明要进行的长期试验的条件。
In general, it is acceptable for stability information to be generated under a more severe climatic zone storage condition already defined in Table 3 to support the labelling. Testing at a more severe long-term condition (e.g., 30°C ±2°C/75% RH ±5% RH) could be justified as it encompasses all climate zones that a drug substance or drug product may be exposed to. However, if it is demonstrated that the drug substance or drug product will not remain within its acceptance criteria when stored at the more severe condition (e.g., 30°C ±2°C/75% RH ±5% RH) for the duration of the proposed re-test period or shelf life, the following are some approaches to consider:
一般而言,在表3中定义的更严酷的气候带贮藏条件下生成的稳定性信息是可以接受的,以支持说明书及标签。在更苛刻的长期条件下(如30°C±2°C/75%RH±5%RH)进行试验是合理的,因为该条件涵盖了原料药或制剂可能暴露的所有气候带。但是,如果证明原料药或制剂在更苛刻的条件下(如30°C±2°C/75%RH±5%RH)在建议的复检期或有效期内贮藏时不能保持在其可接受标准范围内,则应考虑以下方法:
• alternative long-term storage condition for the intended climatic zone.
• 预期气候带的替代长期条件
• a minimal reduction in re-test period or shelf life.
• 复检期或有效期的最小缩减
• evaluation of stability in an alternative container closure system.
• 替代容器密封系统的稳定性评估
• evaluation of formulation and manufacturing process options.
• 剂型和生产工艺方案的评估
When long-term studies are conducted at 25°C ±2°C/60% RH ±5% RH and a significant change occurs at any time during 6 months’ testing under accelerated conditions, additional testing at the intermediate storage condition should be conducted and evaluated against significant change criteria (refer to Section 13 – Data Evaluation).
当在25°C±2°C/60%RH±5%RH条件下进行长期研究,并在加速条件下6个月的试验期间的任何时间发生显著性变化时,应在中间条件下进行额外试验,并根据显著性变化标准进行评估(参见第13节-数据评估)。
If 30°C ±2°C/65% RH ±5% RH or 30°C ±2°C/75% RH ±5% RH is the long-term condition, there is no intermediate condition defined.
如果30°C±2°C/65%RH±5%RH或30°C±2°C/75%RH±5%RH为长期条件,则无需定义中间条件。
For Climatic Zone III stability studies, an alternative approach to studying at the reference relative humidity (e.g., 35% RH ± 5% RH) can be achieved by performing the stability studies under higher relative humidity (e.g., 65% RH ± 5% or 75% RH ± 5%) through mathematical calculation. This can be achieved by experimentally determining the permeation coefficient for the container closure system (e.g., refer to Example 1 in Section 7.2.2 – Storage Conditions for Products Packaged in Semi- Permeable Containers).
对于气候带III 的稳定性研究, 可以通过数学计算在较高相对湿度( 如65%RH±5% 或
75%RH±5%)下进行稳定性研究,从而实现参考相对湿度(如35%RH±5%RH)下的替代研究方法。这可以通过实验确定容器密封系统的渗透系数来实现(例如,参考第7.2.2节“包装在半渗透性容器中的产品的贮藏条件”中的示例1)。
7.2.1 Storage Conditions for Products Packaged in Impermeable Containers
7.2.1 包装在非渗透性容器中的产品的贮藏条件
Since drug substance and drug products packaged in impermeable containers (e.g., aluminium /aluminium foil blister, sealed glass container) provide a permanent barrier to passage of moisture or solvent, sensitivity to moisture or potential for solvent loss is not a concern. Thus, stability studies for products stored in impermeable containers can be conducted under any humidity condition.
由于包装在非渗透性容器(如铝/铝箔泡罩、密封玻璃容器)中的原料药和制剂为水分或溶剂通过提供了永久性屏障,因此对水分的灵敏度或溶剂损失的可能性不是问题。 因此,对于贮藏在非渗透性容器中的产品,可以在任何湿度条件下进行稳定性研究。
7.2.2 Storage Conditions for Products Packaged in Semi-Permeable Containers
7.2.2 包装在半渗透性容器中的产品的贮藏条件
Sensitivity to moisture or potential for solvent loss is a concern for drug substance and drug products packaged in semi permeable containers. Semi-permeable containers can allow the passage of moisture, solvent, or gases while preventing solute loss. The mechanism for solvent transport occurs by absorption into one container surface, diffusion through the bulk of the container material and desorption from the other surface. Transport across the container wall is driven by a partial pressure gradient.
对于包装在半渗透性容器中的原料药和制剂,对水分的灵敏度或溶剂损失的可能性是一个问题。半渗透性容器可以允许水分、溶剂或气体通过,同时防止溶质损失。溶剂运输的机制是通过吸收到一个容器表面、通过容器材料的本体扩散以及从另一个表面解吸而发生的。通过容器壁的运输是由分压梯度驱动的。
Aqueous-based products packaged in semi-permeable containers should be evaluated for potential water loss in addition to physical, chemical, biological and microbiological stability. This evaluation should be carried out under conditions of low relative humidity, as discussed below. Ultimately, it should be demonstrated that aqueous-based products stored in semi-permeable containers can withstand low relative humidity environments.
除物理、化学、生物和微生物稳定性外,还应评估包装在半渗透性容器中的水基产品的潜在失水情况。应在低相对湿度条件下进行该评估,如下所述。最终,应证明贮藏在半渗透性容器中的水基产品能够承受低相对湿度环境。
For non-aqueous, solvent-based products, comparable approaches can be developed and applied.
对于非水性溶剂型产品,可以开发和应用类似的方法。
Table 4: Storage Condition Recommendations for Semi-Permeable Containers
表 4 半渗透性容器的贮藏条件建议
|
Long-term |
Intermediate |
Accelerated |
|
25°C ±2°C/40% RH ±5% RH |
30°C ±2°C/35% RH ±5% RH |
40°C ±2°C/not more than (NMT) 25% RH |
|
30°C ±2°C/35% RH ±5% RH |
Not applicable |
Testing at a more severe long-term condition, e.g., 30°C ±2°C/35% RH ±5% RH could be justified.
|
长期 |
中期 |
加速 |
|
25°C ±2°C/40% RH ±5% RH |
30°C ±2°C/35% RH ±5% RH |
40°C±2°C/NMT 25% RH |
|
30°C ±2°C/35% RH ±5% RH |
不适用 |
在更严苛的长期条件下进行试验是合理的,如30°C±2°C/35%RH±5%RH。
A 5% loss in water from its initial value is considered a significant change for a product packaged in a semi-permeable container after an equivalent of 3 months’ storage at 40°C ± 2°C /NMT 25% RH.However, for small containers (1 mL or less) or unit-dose products, a water loss of 5% or more after an equivalent of 3 months’ storage at 40°C ± 2°C /NMT 25% RH may be acceptable, if justified.
对于包装在半渗透性容器中的产品,在40°C±2°C/NMT 25% RH条件下贮藏3个月后,其水分含量与初始值相比损失5%被视为显著变化。但是,对于小容器(1 mL或更少)或单位剂量制剂,在40°C±2°C/NMT 25% RH条件下贮藏3个月后,失水量≥5%是可以接受的(如果证明合理)。
A significant change in water loss alone under the accelerated condition does not necessitate testing at the intermediate storage condition. However, data should be provided to demonstrate that no significant water loss has been observed throughout the proposed re-test period / shelf life if stored at 25°C ±2°C / 40% RH ±5% RH.
仅在加速条件下失水量发生显著变化时,无需在中间条件下进行试验。但是,应提供数据证明在25°C±2°C/40%RH±5%RH条件下贮藏时,在整个拟议的复检期/有效期内未观察到显著的失水。
When long-term studies are conducted at 25°C ± 2°C/40% RH ± 5% RH, additional testing at the intermediate storage condition should be performed to evaluate the temperature effect at 30°C if significant change other than water loss occurs during the 6 months testing at the accelerated condition.
当在25°C±2°C/40%RH±5%RH条件下进行长期研究时,如果在加速条件下的6个月试验期间发生失水以外的显著变化,则应在中间条件下进行额外试验,以评估30°C下的温度影响。
If 30°C ±2°C/35% RH ±5% RH is the long-term condition, there is no intermediate condition.
如果长期条件为30°C±2°C/35%RH±5%RH,则无需设置中间条件。
An alternative approach to performing studies at the reference relative humidity as recommended in Table 5 is performing the stability studies under higher relative humidity and deriving the water loss at the reference relative humidity through calculation. This can be achieved by experimentally determining the permeation coefficient for the container closure system (e.g., as shown in the illustrative example below, using the calculated ratio of water loss rates for the container closure system between the two relative humidity conditions at the same temperature). The permeation coefficient for a container closure system can be experimentally determined by using the worst-case scenario (e.g., the most diluted of a series of concentrations) for the proposed drug product.
在表5中推荐的参考相对湿度下进行研究的另一种方法是在较高相对湿度下进行稳定性研究,并通过计算得出在参考相对湿度下的失水量。这可以通过实验确定容器密封系统的渗透系数来实现(例如,如以下说明性示例所示,通过计算在相同温度下两种相对湿度条件下容器密封系统的失水率的比率)。容器密封系统的渗透系数可以通过使用拟申报制剂的最差情况(例如,一系列浓度的最大稀释度)进行实验确定。
Example 1. An approach for determining water loss:
示例1测定失水的方法:
For a product in a given container closure system, container size and fill, an appropriate approach for deriving the water loss rate at the reference relative humidity is to multiply the water loss rate measured at an alternative relative humidity at the same temperature by a water loss rate ratio determined experimentally shown in Table 5 below. A linear water loss rate at the alternative relative humidity over the storage period should be demonstrated.
对于在特定容器密封系统、容器尺寸和装量的产品,推导参考相对湿度下的失水率的适当方法是将相同温度下在替代相对湿度下测得的失水率乘以通过实验确定的失水率比(见下表5)。应证明贮藏期间在替代相对湿度下的线性失水率。
For the below illustrative example, at a given temperature (e.g., 40°C) the water loss rate determined experimentally for the proposed container closure system during storage at NMT 25% RH is the water loss rate measured at 75% RH multiplied by 3.0, the corresponding water loss rate ratio.
对于下面的说明性示例,在设定的温度(例如40°C)下,通过实验确定的拟定的容器密封系统在不超过25%RH下贮藏期间的失水率是在75%RH下测得的失水率乘以3.0,即相应的失水率比。
Table 5: Example of Ratio of Water Loss Calculations
表 5失水率计算示例
|
Alternative relative humidity |
Reference relative humidity |
Ratio of water loss rates at a given temperature1 |
|
60% RH |
25% RH |
1.9 |
|
60% RH |
40% RH |
1.5 |
|
65% RH |
35% RH |
1.9 |
|
75% RH |
25% RH |
3.0 |
|
1Ratio of water loss = (100 - Reference % RH)/(100 - Alternative % RH) |
||
|
替代相对湿度 |
参考相对湿度 |
给定温度下的失水率比 1 |
|
60% RH |
25% RH |
1.9 |
|
60% RH |
40% RH |
1.5 |
|
65% RH |
35% RH |
1.9 |
|
75% RH |
25% RH |
3.0 |
1失水率= (100 - 参考%RH) / (100 - 替代%RH)
The ratios described in Table 5 above are for illustrative purposes. Actual ratios for water loss rates determined experimentally for the proposed container closure system under various relative humidity conditions should be provided.
上述表5中描述的比率仅用于说明目的。应提供在各种相对湿度条件下,通过实验测定的拟定容器密封系统失水率的实际比率。
7.3 Considerations for Refrigerated Temperature Storage
7.3 冷藏温度贮藏的注意事项
Recommendations for drug substance and drug products intended for long-term storage under
refrigerated conditions are provided below. Accelerated conditions are intended to demonstrate the effect of temperature, and active humidity control may not be needed when justified.
对拟在冷藏条件下长期贮藏的原料药和制剂的建议如下。加速条件旨在证明温度和主动湿度控制的影响,如果合理,可能不需要。
Table 6: Storage Under Refrigerated Conditions
表 6 冷藏条件下的贮藏
|
Long-term |
Accelerated |
|
5°C ±3°C |
25°C ±2°C or any alternative temperature condition when justified. |
|
长期 |
加速 |
|
5°C ±3°C |
25°C±2°C或任何替代温度条件(如合理)。 |
For an aqueous-based product packaged in a semi-permeable container, appropriate information should be provided to assess the extent of water loss.
对于包装在半渗透性容器中的水性产品,应提供适当的信息以评估失水程度。
For products stored under refrigerated conditions, when a significant change or out of specification occurs within the first 3 months of testing under accelerated conditions, a discussion should be provided to address the effect of shipment and handling (refer to Section 14 – Labelling).
对于在冷藏条件下贮藏的产品,当在加速条件下试验的前3个月内发生显著变化或超标时,应进行讨论以说明运输和搬运的影响(参见第14节 - 说明书及标签)。
For synthetics, it is considered unnecessary to continue to test a product under accelerated conditions through 6 months when a significant change has occurred within the first 3 months.
对于合成制剂,当在前3个月内发生显著变化时,则认为无必要在加速条件下继续检测产品直至6个月。
7.4 Considerations for Frozen Temperature Storage
7.4 冷冻温度贮藏的注意事项
Recommendations for drug substance and drug products intended for long-term storage under frozen conditions (as determined for the product) are provided below.
对拟在冷冻条件下长期贮藏的原料药和制剂(根据产品确定)的建议如下。
Table 7: Storage in a Freezer or below -20°C
表 7 冷冻柜或-20°C 以下贮藏
|
Long-term |
|
-20°C or below |
|
长期 |
|
-20°C或以下 |
Testing at accelerated or stress conditions (e.g., 5°C ± 3°C or 25°C ± 2°C or 30°C ± 2°C or any appropriate condition based on the intrinsic properties of the drug substance or drug product) for an appropriate time period should be conducted to address the effect of short-term excursions outside the proposed label storage condition (refer to Section 14.1- Excursions Outside of a Labelling Claim).
应在加速或下(如5°C±3°C、25°C±2°C、30°C±2°C或基于原料药或制剂内在特性的任何适当条件)进行适当的时间周期的试验,以研究短期偏移拟定说明书及标签贮藏条件的影响(参见第14.1节 - 说明书及标签声明之外的偏移)。
8 PHOTOSTABILITY
8 光稳定性
8.1 Purpose of Photostability Testing
8.1 光稳定性试验的目的
This section addresses the principles governing the generation of photostability information in initial regulatory submission and for lifecycle management changes.
本节介绍了初始注册申报和全生命周期管理中的中光稳定性信息的指导原则。
The intrinsic photostability characteristics of a product should be evaluated to demonstrate that light exposure does not result in unacceptable change that could compromise product efficacy or patient safety. Normally, photostability testing is carried out on a single representative batch suitable for the purpose of the study. Repeating a photostability study may be required in response to relevant changes (e.g., in the formulation, container closure system and in-use conditions) when the photostability characteristics and controls established at the time of the initial regulatory submission are assessed to be impacted (refer to Section 15.3 – Product Lifecycle Stability Studies).
应评估产品的固有的光稳定性特性,以证明光暴露不会导致可能危及产品有效性或患者安全性的不可接受的变化。通常,光稳定性试验是在适合研究目的的单个代表性批次上进行的。当评估初始监管申报时建立的光稳定性特征和控制措施会受到影响时,可能需要重复光稳定性研究以应对相关变化(例如剂型、容器密封系统和使用条件的变化)(参见第15.3节-产品生命周期稳定性研究)。
Two specific studies are performed to generate and evaluate photostability data:
为生成和评估光稳定性数据,进行了两项特定研究:
Forced photodegradation study – A study that may be an integral part of forced degradation evaluation and may be undertaken in the development phase. This information may be used to evaluate the overall photosensitivity of the drug substance and drug product for method development purposes, degradation pathway elucidation and to inform control strategies (refer to Section 2-Development Stability Studies Under Stress and Forced Conditions).
• 强制光降解研究 - 可能是强制降解评估的一个组成部分,并可在开发阶段进行。该
信息可用于评估原料药和制剂的整体光敏性,用于方法开发、降解途径阐明和控制策略(参见第2节 - 影响因素和强制降解条件下的开发稳定性研究)。
Confirmatory photostability studies – Studies performed when a risk of photodegradation has been identified. The purpose of the studies is to establish the photostability characteristics to understand the ability of the primary or secondary packaging material to protect light-sensitive products and the impact of light on product quality through manufacture, storage, transportation and in-use. These data may also support labelling (e.g., storage statements).
• 确认光稳定性研究 - 当确定存在光降解风险时进行的研究。研究的目的是确定光稳
定性特性,了解内包装或外包装材料在生产、贮藏、运输和使用过程中保护光敏感产品的能力以及光对产品质量的影响。这些数据也可支持说明书及标签信息(例如,存储说明)。
A systematic approach to photostability testing is recommended, covering as appropriate:
建议采用系统方法进行光稳定性试验,包括(如适用):
i) Tests on the drug substance and/or drug product directly exposed; and if necessary.
i) 对直接暴露的原料药和/或制剂的检测;如有必要。
ii) Tests on the drug substance and/or drug product in the primary packaging; and if necessary.
ii) 对内包装中的原料药和/或制剂的检测;如有必要。
iii) Tests on the drug substance and/or drug product in the secondary packaging.
iii) 对外包装中原料药和/或制剂的检测。
Normally, the studies are carried out in a sequential manner starting with testing the sample directly exposed then progressing as necessary to the drug substance and/or drug product in the primary packaging and then in the secondary packaging, if applicable. If the product is known to be photosensitive, e.g., most biologicals, parallel testing can be carried out as a science- and risk -based approach. The extent of testing should be established by assessing whether acceptable change or no change has occurred at the end of the light exposure testing. Acceptable change is a change within limits previously justified by the applicant. If a non-acceptable change is observed, a change in the packaging or the formulation should be proposed. Testing should progress until the results demonstrate that the drug substance and/or drug product is adequately protected from exposure to light (refer to Figure 3 - Decision Flow Chart for Systematic Photostability Testing).
通常情况下,研究是按顺序进行的,首先测试直接暴露的样品,然后根据需要对内包装中的原料药和/或制剂进行检测,然后对外包装中的原料药和/或制剂进行检测(如适用)。如果已知产品(例如大多数生物制品)具有光敏性,则可以基于科学和风险进行平行试验。应通过评估光暴露试验结束时是否发生可接受的变化或无变化来确定试验范围。可接受的变化是指在申请人先前证明的限度范围内的变化。如果观察到不可接受的变化,应提议对包装或剂型进行变更。试验应持续进行,直至结果证明原料药和/或制剂充分避光(参见图3 - 系统光稳定性试验的决策流程图)。
Figure 3: Decision Flow Chart for Systematic Photostability Testing
图 3 系统光稳定性试验的决策流程图
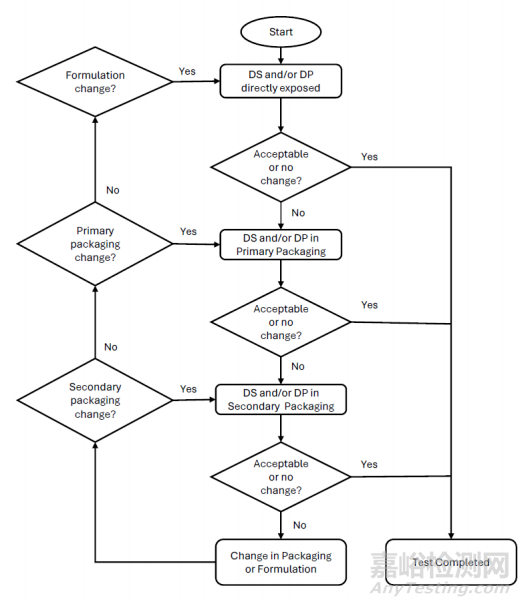
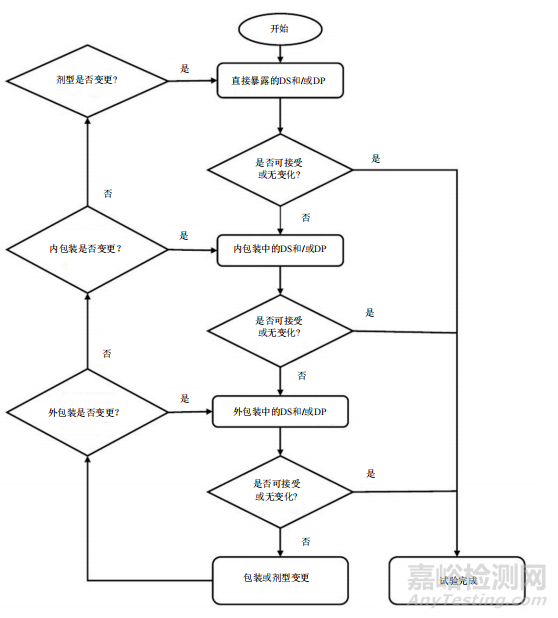
8.2 Forced Photodegradation
8.2 强制光降解
As forced photodegradation is an integral part of forced degradation strategy, details on the concepts,study design considerations and interpretation of results can be found in Section 2- Developmental Studies Under Stress and Forced Conditions. For details on radiation sources and light exposure conditions for forced photo degradation studies refer to Section 8.4 – Radiation Source and Light Exposure.
由于强制光降解是强制降解策略的组成部分,有关概念、研究设计注意事项和结果分析的详细信息,请参见第2节 – 影响因素和强制降解条件下的开发研究。有关强制光降解研究的辐射源和光暴露条件的详细信息,请参见第8.4节 - 辐射源和光暴露。
If the forced photodegradation study is combined with the confirmatory photostability study, the specific sample considerations provided in Section 8.3 - Confirmatory Photostability should be considered, e.g., for solid substances.
如果结合强制光降解研究与确认光稳定性研究,应考虑第8.3节 - 确认光稳定性中提供的特定样本考虑因素,例如对于固体物质。
8.3 Confirmatory Photostability
8.3 确认光稳定性
The confirmatory studies are used to determine whether special precautionary measures are needed in manufacturing, formulation of the product, long-term storage or in-use period (refer to Section 11 - In-Use Stability) and if a light-resistant container closure system and/or special labelling information are needed. Guidance is provided on determining whether a confirmatory study should be performed, study design and interpretation of results (refer to Figure 3- Decision Flow Chart for Systematic Photostability Testing).
确认研究用于确定在生产、产品剂型、长期贮藏或使用期间是否需要采取特殊预防措施(参见第11节 - 使用中稳定性),以及是否需要避光容器密封系统和/或特殊说明书及标签信息。为确定是否应进行确认研究、研究设计和结果分析提供了指导(参见图3 - 系统光稳定性试验的决策流程图)。
For synthetic chemical entities, confirmatory photostability testing is generally performed on one batch of the drug substance and the drug product, while for biologicals, testing is generally performed on one batch of the drug product. Confirmatory testing is typically conducted in the primary container closure system and including, if necessary, secondary packaging. Alternative science- and risk-based approaches may be considered when appropriately justified and may include scenarios where confirmatory photostability testing is not required. For example, if no photodegradation is observed in the fully exposed drug substance sample or the fully exposed drug product sample, no further testing as part of the confirmatory study is needed. For some products where it has been demonstrated that the
primary packaging is completely impenetrable to light (e.g., aluminium tubes cans or foil/foil blisters) testing should normally only be conducted on directly exposed drug product.
对于化学合成实体,通常对一批原料药和制剂进行确认光稳定性试验,而对于生物制品,通常对一批制剂进行试验。通常在内容器密封系统中进行确认试验,如有必要,包括外容器密封系统。在适当证明合理时,可以考虑替代科学和基于风险的方法,并且可能包括无需进行确认光稳定性试验的情况。例如,如果在完全暴露的原料药样品或完全暴露的制剂样品中未观察到光降解,则无需作为确认研究一部分进行进一步试验。对于某些已证明内包装完全不透光的产品(如铝管、罐或箔/箔泡罩),通常仅对直接暴露的制剂进行试验。
If the results from the confirmatory study batch are not conclusive in terms of photostability or photolability, testing of additional batches or a new study design should be considered.
如果确认研究批次的结果在光稳定性或光敏性方面无法得到结论,则应考虑对其他批次进行试验或采用新的研究设计。
As a direct challenge for samples of solid products, an appropriate amount of sample should be taken and placed in a glass or plastic dish spread in a single layer and protected with a suitable transparent cover, if considered necessary. Tablets and capsules should be spread in a single layer. Solids, except tablets or capsules, should be spread across the dish to give a thickness of typically not more than 3 millimetres. When direct exposure is not feasible (e.g., liquids, or products sensitive to non-light induced oxidation), the sample should be placed in a suitable protective inert transparent container (e.g., quartz). In general, the samples should be positioned to provide maximum area of exposure to the light source.
作为对固体产品样品的直接挑战,应采集适量的样品,并将其放置在单层铺开的玻璃或塑料皿中,并在必要时用适当的透明盖保护。片剂和胶囊应单层铺开。固体样本(除片剂或胶囊外)应在培养皿上铺开,厚度通常不超过3毫米。当直接暴露不可行时(例如,液体或对非光致氧化敏感的产品),样品应放置在合适的保护性惰性透明容器中(如石英)。一般而言,样品应放置在能提供最大光源区域面积的位置。
If testing of the drug product in the primary or secondary packaging is needed, the samples should be placed horizontally or transversely with respect to the light source, providing the most uniform exposure of the samples. Some adjustment of testing conditions may have to be made when testing large-volume containers (e.g., dispensing packs). In general, samples with the greatest light exposure surface in the container should be tested.
如果需要对内包装或外包装中的制剂进行检测,样品应相对于光源水平或横向放置,以使样品暴露最均匀。在测试大体积容器(例如,分发包装)时,可能需要对测试条件进行一些调整。一般而言,应检测容器中受光面最大的样品。
At the end of the exposure period, representative samples (taking homogeneity of light exposure into consideration) should be examined by analytical procedures (suitable for intended purpose) for any changes in physical, chemical or biological properties, including assay or potency and degradants that are determined from the characterisation studies that are likely to arise from photochemical degradation.
When powder samples are involved, sampling should ensure that a representative portion is used in individual tests. For solid oral dosage form products (e.g., tablets, capsules), testing should be conducted on a suitable number of units (statistical sampling approaches may be used). Similar sampling considerations, such as homogeneity or solubilisation of the entire sample, apply to other materials that may not be homogeneous after exposure (e.g., creams, ointments, suspensions).
在暴露期结束时,应采用分析方法(适用于预期目的)检查代表性样品(考虑光暴露的均一性),以确定物理、化学或生物特性、包含含量或效价以及降解物的任何变化。这些变化由特性鉴定研究确定,可能由光化学降解引起。当涉及粉末样品时,取样应确保在单个试验中使用具有代表性的部分。对于固体口服剂型产品(如片剂、胶囊),应在适当数量的单位上进行检测(可采用统计抽样方法)。类似的取样考虑因素,如整个样品的均匀性或溶解性,适用于暴露后可能不均匀的其他材料(如乳膏、软膏、混悬液)。
暴露样品的分析应与用作暗对照的任何受保护样品(如果在试验中使用)的分析同时进行。
The analysis of the exposed sample should be performed concomitantly with that of any protected samples used as dark controls if these are used in the test. When evaluating the results of photostability studies to determine whether change due to exposure to light is acceptable, it is important to consider the results obtained from other formal stability studies to assure that the product will be within proposed specifications during the re-test period or shelf life. Depending on the extent of change or failure to meet acceptance criteria, special precautions may be needed to mitigate exposure to light, like formulation change, redesign of container closure system (including secondary packaging), a reduced re-test period or shelf life of drug substance or drug product (in conjunction with long term stability data) or change in labelling for storage and use (refer to Figure 3 - Decision Flow Chart for Systematic Photostability Testing).
在评价光稳定性研究结果以确定是否可以接受光暴露引起的变化时,重要的是要考虑从其他正式稳定性研究中获得的结果,以确保产品在复检期或有效期内符合拟定的质量标准。根据变化程度或不符合可接受标准的情况,可能需要采取特殊的预防措施来减少光暴露,如剂型变更、重新设计容器密封系统(包括外包装)、缩短原料药或制剂的复检期或有效期(结合长期稳定性数据)或贮藏和使用说明书及标签的变更(参见图3 - 系统光稳定性试验的决策流程图)。
8.4 Radiation Source and Light Exposure
8.4 辐射源和光暴露
This section describes the radiation source and light exposure that can be used to support forced photodegradation studies and confirmatory photostability studies. For forced degradation studies a variety of exposure conditions may be used, depending on the photosensitivity of the product and the intensity of the light sources used. Confirmatory photostability studies should be based on light exposure possible during manufacture, storage, distribution and in-use.
本节描述了可用于支持强制光降解研究和确认光稳定性研究的辐射源和光暴露。对于强制降解研究,可以使用各种暴露条件,具体取决于产品的光敏性和所用光源的强度。确认光稳定性研究应基于生产、贮藏、运输和使用期间可能的光暴露。
In photostability studies, it is important to consider the spectral characteristics of the light, cumulative light exposure and temperature, as the combination of these factors will influence the rate of photodegradation and the design of the study.
在光稳定性研究中,重要的是考虑光的光谱特征、累积光暴露和温度,因为这些因素的组合将影响光降解速率和研究设计。
The light sources described below are considered appropriate for photostability testing. Alternative light sources may be applicable when justified. The applicant should either maintain appropriate temperature control to minimise the effect of localised temperature changes or include a dark control in the same environment unless otherwise justified. The applicant may rely on the spectral distribution specification of the light source manufacturer for the following options:
下述合适的光源可用于光稳定性试验。如合理,可采用替代光源。除非另有说明,否则申请人应保持适当的温度控制,以尽量减少局部温度变化的影响,或在相同环境中设置暗对照。申请人可根据光源制造商的光谱分布规范选择以下方案:
Option 1:
选项 1:
For light exposure similar to the D65 (outdoor daylight) emission standard (as currently defined in,ISO/CIE 18909:2022) (17), an artificial daylight fluorescent lamp combining visible and ultraviolet(UV) outputs, xenon or metal halide lamp, including appropriate filter(s) is recommended as radiation light source.
对于类似于D65(室外日光)排放标准(目前定义见ISO/CIE 18909:2022)(17)的光暴露,建议使用结合可见光和紫外线(UV)输出的人造日光荧光灯、氙气或金属卤化物灯,包括采用适当的滤光器作为辐射光源。
Option 2:
选项 2:
A combined exposure to both cool white fluorescent and near ultraviolet lamp, which is capable of producing a light exposure similar to the ID65 (indoor daylight) emission standard, for which the ultraviolet lamp has at least 25% of the ultraviolet-A between 320 and 360 nm and at least 25% is between 360 and 400 nm.
组合暴露于冷白色荧光灯和近紫外线灯,其能够产生类似于ID65(室内日光)发射标准的光暴露,对于该标准,紫外线灯在320 nm和360 nm之间具有至少25%的UV-A并且至少25%是在360和400nm之间。
Option 3:
选项 3:
Ambient/mild light conditions (predominantly light >400 nm during manufacturing, processing and in- use), for which a fluorescent or LED lamp is recommended.
采用环境/温和光照条件(制造、加工和使用过程中的主要光线>400 nm),建议使用荧光灯或LED灯。
Light exposure for forced photodegradation studies may require higher light intensity, such as doubling the levels used in confirmatory studies. However, depending on the photosensitivity of the product, milder conditions may be more suitable to avoid extensive decomposition. For example, samples might be exposed to ambient/mild light conditions, typically ranging from 43-260 ×103 lux hours for >400 nm and 0.3-3 Wh/m2 for 350 – 400 nm, over an exposure period of 1 to 7 days.
强制光降解研究的光暴露可能需要更高的光强度,例如将确认研究中使用的光强度水平加倍。然而,根据产品的光敏性,较温和的条件可能更为适宜,以避免过度分解。例如,样品可能暴露在环境/温和的光照条件下,暴露时间为1至7天,通常范围为43-260×103勒克斯小时(>400 nm)和0.3-3 Wh/m2(350-400 nm)。
In confirmatory studies, to assess the effects of light under controlled conditions during manufacturing, storage and in use, samples maybe exposed to light providing an overall illumination of not less than 1.2 million lux hours and an integrated near ultraviolet energy of not less than 200 Wh/m2. When justified, alternate approaches may also be appropriate depending on the photosensitivity of the product, the light source selected, manufacturing conditions and packaging. The overall light exposure during manufacture can be determined by measuring the light exposure and defining the average light exposure and UV energy (e.g., in Luxh and/or Wh/m2). The average light exposure reading, with the worst-case light exposure time, could be used to define light exposure time and distance to light source
considerations in the confirmatory study.
在确认研究中,为了评估在生产、贮藏和使用过程中受控条件下的光照影响,可将样品暴露于总照度不低于120万勒克斯小时且综合近紫外能量不低于200 Wh/m2的光照下。在合理的情况下,根据产品的光敏性、选择的光源、制造条件和包装,也可采用合适的替代方案。可以通过测量光暴露并定义平均光暴露和UV能量(例如,以Luxh和/或Wh/m2为单位)来确定制造过程中的总体光暴露。在确认性研究中,可使用最差情况下的光照暴露时间的平均光照读数来界定光照暴露时间和与光源的距离。
9 STABILITY CONSIDERATIONS FOR PROCESSING AND HOLDING TIMES FOR INTERMEDIATES
中间产品加工和贮藏时间的稳定性考虑因素
General Considerations
9.1 一般考虑因素
Good manufacturing practices (GMP) and good distribution practices (GDP) require that controls are in place to ensure that intermediates (i.e., drug substance intermediates and drug product intermediates(including bulk drug products)) are manufactured and stored under appropriate conditions. Storage and/or transportation arrangements should not have deleterious effects on the subsequent processing,stability, safety, or quality of intermediates, in accordance with good distribution practices.
药品生产质量管理规范(GMP)和良好分销规范(GDP)要求采取控制措施,以确保在适当的条件下生产和贮藏中间产品(即原料药中间产品和制剂中间产品(包括散装制剂))。根据良好分销规范,贮藏和/或运输安排不应对中间产品的后续加工、稳定性、安全性或质量产生不利影响。
The processing time can be considered as the established time period needed to perform a manufacturing step or series of steps and should take into consideration compatibility with manufacturing equipment.Whereas the holding time can be considered as the established time period for which materials (e.g.,dispensed raw materials, drug substance intermediates and drug product intermediates) are awaiting further processing or packaging in the final container closure system and may be held and/or transported under specified conditions. For such intermediates, maximum holding times should be established to ensure their quality and that they can be held, pending the next processing step, without having results outside the established control strategy. Intermediates should not be used beyond the established holding times. A written protocol, procedure or program for the holding time studies should be followed taking into consideration the principles described in Section 3.1 – General Principles.
加工时间可视为执行一个生产步骤或一系列步骤所需的既定时间段,并应考虑与生产设备的相容性。鉴于贮藏时间可视为物料(例如,分配的原料、原料药中间产品和制剂中间产品)在最终容器密封系统中等待进一步加工或包装并可在特定条件下保存和/或运输的既定时间段。对于此类中间产品,应确定最长贮藏时间,以确保其质量,并确保其可贮藏至下一个加工步骤之前,而不会产生超出既定控制策略的结果。中间产品的使用不应超过规定的贮藏时间。考虑到第3.1节 - 一般原则中描述的原则,应遵循贮藏时间研究的书面方案、规程或计划。
The data used to establish the holding time should cover the proposed holding times for the
intermediates and the stability studies should be performed at relevant temperature and humidity conditions to support the expected storage conditions for the drug substance or drug product intermediate. If the temperature and humidity conditions used during these studies do not correspond with the storage conditions described in Section 7 - Storage Conditions of this guideline, other conditions should be justified. If the product is sensitive to light exposure that may occur during storage, data should confirm that controls are sufficient to limit exposure to acceptable levels as described in Section 8 - Photostability. If more than one production site is involved, the stability studies should also consider transportation of the intermediates. For consideration of reduced design, the principles of Annex 1 - Reduced Stability Protocol Design may apply. Cumulative hold times are generally assessed as part of process validation. If a stability risk is identified, a cumulative holding time study may be necessary.
用于确定贮藏时间的数据应涵盖中间产品的建议贮藏时间,稳定性研究应在相关温度和湿度条件下进行,以支持原料药或制剂中间产品的预期贮藏条件。如果这些研究中使用的温度和湿度条件与本指导原则第 7 节 “贮藏条件”中描述的贮藏条件不一致,则应证明其他条件的合理性。如果产品对贮藏期间可能发生的光暴露敏感,则应提供数据确认控制措施足以将光暴露限制在可接受水平,如第8节 - 光稳定性中所述。如果涉及多个生产场地,稳定性研究还应考虑中间产品的运输。考虑简化设计时,附录1-简化稳定性方案设计的原则可能适用。累积贮藏时间通常作为工艺验证的一部分进行评估。如果发现稳定性风险,可能需要进行累积贮藏时间研究。
For drug substance and drug product intermediates that are packaged and stored outside of the
manufacturing process activities or that are purchased as such, it may be appropriate to establish a re-test period or shelf life, as applicable, rather than a holding time. In these situations, the recommendations described in the respective sections within this guideline should be followed for the stability studies conducted to support the re-test period or shelf life with the corresponding storage statements.
对于在生产工艺活动之外包装和贮藏的原料药和制剂中间产品,或以这种方式购买的原料药和制剂中间产品,可能适合确定复检期或有效期(如适用),而非贮藏时间。在这些情况下,稳定性研究应遵循本指导原则相应章节中所述的建议,以支持复检期或有效期以及相应的存储说明。
Stability recommendations for intermediates, including considerations that are specific for synthetic chemical entities and biologicals, are described below.
关于中间产品的稳定性建议,包括针对化学合成实体和生物制品的特定考虑因素,如下所述。
Considerations for Synthetic Chemical Entities
9.2 化学合成实体的考虑因素
The holding times of the drug substance intermediates should consider GMP principles and comply with written procedures. However, in situations where an in-process step for the drug substance has a holding time where the quality of the drug substance may be affected by the hold, then the principles in this section apply.
原料药中间产品的保持时限应考虑GMP原则,并遵循书面规程。但是,如果原料药的中间过程步骤有保持时限规定,而该保持时限可能会影响原料药的质量,则本节中的原则适用。
When established, the processing times and maximum holding times for drug product intermediates should be included in the description of the manufacturing processes. The risk assessment and control strategy for the drug product manufacturing processes should include an assessment of whether holding time studies should be performed. When applicable, the information to support the processing and holding times should be included in the regulatory submission.
制剂中间产品的加工时间和最长保持时限一经确定,应包含在生产工艺描述中。制剂生产工艺的风险评估和控制策略应包括是否应进行保持时限研究的评估。如适用,应在注册申报资料中纳入支持加工和保持时限的信息。
When the holding times of a drug product intermediate are prolonged (e.g., more than 30 days for solid dosage forms for the entire manufacturing process or more than 24 hours for non-solid dosage forms or sterile products), evidence of the suitability of the holding times, together with the proposed container that is representative of that for marketing, the storage period or transportation arrangements, should be included in the regulatory submission, when requested. Where intermediates are transported between production sites, the transportation arrangements and method of transportation should be described in general terms (e.g., intermediate container, storage and transportation conditions) in the description of the manufacturing processes.
当制剂中间产品的保持时限延长时(例如,在整个生产过程中固体剂型超过 30 天,或非固体剂型或无菌产品超过 24 小时),应根据要求在注册申报资料中纳入保持时限的适用性证据,以及代表上市包装的拟定容器、贮藏期或运输安排。当在生产场地之间运输中间产品时,应在制造工艺描述中以通用术语(例如中间产品容器、贮藏和运输条件)描述运输安排和运输方法。
For a drug substance or drug product produced by batch processes (i.e., not by continuous
manufacturing processes), it is expected that the data to support the holding times is generated and is representative of the overall process. If the data to support the holding times were not generated on production scale batches, these data should be verified in post-approval stability commitment to conduct these studies on production scale batches. If continuous manufacturing processes are used, the principles outlined in ICH Q13 guideline should be followed when selecting batches to support holding times.
对于通过分批工艺(即,非连续生产工艺)生产的原料药或制剂,预期会生成支持保持时限的数据,并代表整个工艺。如果生产规模批次没有生成支持保持时限的数据,则应在批准后稳定性承诺中确认这些数据,以对生产规模批次进行这些研究。如果使用连续生产工艺,在选择批次以支持保持时限时,应遵循ICH Q13指导原则中概述的原则。
Considerations for Biologicals
9.3 生物制品的注意事项
During the manufacture of biologicals, the quality and control of certain process intermediates may be critical to the production of the drug substance or drug product. In general, the manufacturer should identify process intermediates and generate data and define process limits and holding times that assure their stability within the conditions of the developed manufacturing process. Samples are periodically tested for product quality attributes that may be affected by the holding time.
在生物制品生产过程中,某些工艺中间产品的质量和控制可能对原液或制剂的生产至关重要。一般而言,生产商应确定工艺中间产品并生成数据,并定义工艺限度和保持时限,以确保其在已开发的生产工艺条件下的稳定性。定期检测样品可能受保持时限影响的产品质量属性。
A holding time study for a biological will typically consider two elements: (a) physicochemical stability and (b) microbial control strategy. The physicochemical stability part may be performed on small scale batches that are representative of production scale as part of process characterisation and should be assessed by monitoring relevant CQAs, such as purity and impurity. Microbial control should be demonstrated for the manufacturing process of production scale batches. The use of surrogate material as well as other approaches should be justified.
生物制品的保持时限研究通常会考虑两个因素:(a)理化稳定性和(b)微生物控制策略。作为工艺表征的一部分,理化稳定性部分可在代表生产规模的小规模批次中进行,并应通过监测相关CQA(如纯度和杂质)进行评估。应对生产规模批次的生产工艺进行微生物控制验证。应证明使用替代材料以及其他方法的合理性。
When physicochemical and microbial hold times are determined from separate studies, the established hold time would be the shorter of the two times.
当理化和微生物保持时限由不同的研究确定时,确定的保持时限应为两者中的较短者。
When analytical procedures cannot be applied to an intermediate to determine its holding time, the adequacy of the holding time could be supported by evaluating the quality of the later stage intermediates, drug substance, or drug product.
当无法用分析方法来确定中间产品的保持时限时,可通过评估后期中间产品、原液或制剂的质量来支持保持时限的充分性。
Examples of Holding Time Risk Assessment Considerations
9.4 保持时限风险评估考虑因素示例
The following are examples of the stages that may be considered during the risk assessment of two different types of drug product manufacturing process. Depending on the dosage form, other stages and considerations could be relevant.
以下是两种不同类型的制剂生产工艺风险评估期间可能考虑的阶段示例。根据剂型的不同,其他阶段和考虑因素也可能相关。
Non-Sterile, Solid Oral Dosage Form
非无菌固体口服剂型
The following are examples of the stages that may be considered during the risk assessment of the drug product manufacturing processes for a for a non-sterile, solid oral dosage form to identify potential processing and holding times for intermediates. Depending on the dosage form, other stages and considerations could be relevant.
以下是非无菌固体口服剂型的制剂生产工艺风险评估期间可能考虑的阶段示例,以确定中间产品的潜在加工和保持时限。根据剂型的不同,其他阶段和考虑因素也可能相关。
Table 8: Production steps and associated intermediates for non-sterile, solid oral dosage form
表 8非无菌固体口服剂型的生产步骤和相关中间产品
|
Production Step |
Intermediate |
|
Binder preparation to granulation |
Granulate |
|
Wet granulation to drying |
Dried granulate |
|
Dried granules to lubrication/blending |
Lubricated blend |
|
Mixing to a dry blend |
Blend |
|
Granulation to compressed tablets |
Tablet Cores |
|
Coating solution/suspension to preparation |
Coating solution/suspension |
|
Coating to packaging in bulk containers |
Bulk coated tablets |
|
生产步骤 |
中期 |
|
制粒用粘合剂制备 |
颗粒 |
|
湿法制粒至干燥 |
干燥颗粒 |
|
干燥颗粒至润滑/混合 |
润滑混合物 |
|
混合至干混合物 |
混合 |
|
制粒至压片 |
片芯 |
Sterile, Injectable Solution
灭菌的,注射用溶液
The following are examples of the stages that may be considered during the risk assessment of the manufacturing processes for a sterile, injectable solution to identify potential processing and holding times for intermediates:
以下是灭菌的,注射用溶液生产工艺风险评估期间可能考虑的阶段示例,以确定中间产品的潜在加工和保持时限:
Processing times at 15-25°C during drug substance process to bulk drug substance
• 原料药在15-25°C下分散溶解的工艺时间
Frozen in-process materials
• 工艺中物料的冷冻
Processing time at room temperature (e.g., 15-25°C) from start of drug product manufacturing(e.g., drug substance thaw) until end of fill
• 室温(如15-25°C)下从制剂生产开始(如原料药解冻)至灌装结束的工艺时间
10 SHORT-TERM STORAGE CONDITIONS
10 短期贮存条件
The drug product labelling (refer to Section 14 – Labelling) may specify a short-term storage condition for a drug product. Short term storage is a condition where the primary container closure is not breached and that is different form the long-term storage condition and the in-use period. The short-term storage condition does not need to be implemented by the patient/health care professional, as use of short-term storage is optional. The short-term storage condition is intended for convenience of the patient or health care professional in accordance with regional requirements based on anticipated storage of the drug product. For example, a short-term storage condition would enable a patient to store a refrigerated drug
product at a room temperature condition for a specified duration of time. In these cases, the short-term storage condition and duration should be stated on the labelling along with the long-term storage condition and shelf life. The short-term storage condition is not intended to be applied beyond the shelf life of the drug product. The short-term storage condition is different from any necessary manipulation(e.g., equilibration to ambient temperature) that would be required to prepare a drug for administration(e.g., as per relevant instructions in Instructions for Use). If the drug product can be returned to long-term storage conditions after an acceptable period of short-term storage, data to support the short-term storage conditions should be provided as part of the primary stability studies. A short-term storage
condition is not required for all products. Once a short-term storage condition is established it does not need to be reevaluated periodically unless there is a change likely to impact stability.
制剂说明书及标签(参见第14节 - 说明书及标签)可规定制剂的短期条件。短期贮存条件是指主容器密封系统未被破坏的情况,且与长期贮存条件和使用期允许时限不同。短期贮存条件无需由患者/医护人员实施,因为短期贮存条件是可选择的。根据制剂预期贮藏的区域要求,短期贮存条件旨在方便患者或医护人员。例如,短期贮存条件将使患者能够在室温条件下贮藏冷藏制剂指定持续时间。在这些情况下,应在标签上注明短期贮存条件和持续时间以及长期贮存条件和有效期。短期贮存条件不适用于超过有效期的制剂。短期贮存条件不同于制备给药用药物所需的任何必要操作(如平衡至环境温度)(如按照使用说明书及标签中的相关说明)。如果制剂在经过可接受的短期贮存期后可恢复至长期贮存条件,则应在注册稳定性研究中提供支持短期贮存条件的数据。并非所有产品都需要短期贮存条件。短期贮存条件确定后,无需定期重新评估,除非发生可能影响稳定性的变化。
The design of specific short-term storage condition stability studies should follow the general principles applied to long-term stability studies (refer to Section 3 – Stability Protocol Design) and should consider all relevant climatic zones. Generally, a minimum of 2 batches should be included in the study. The number of batches and the considerations for aged sample should be based on the general principles described for in-use stability studies (refer to Section 11.2.1 – Selection of Batches). Additionally, the applicant may justify alternative strategies, such as modelling (refer to Annex 2 – Stability Modelling), to support the short-term storage condition.
特定的短期贮存条件稳定性研究的设计应遵循适用于长期稳定性研究的一般原则(参见第3章-稳定性方案设计),并应考虑所有相关气候带。一般来说,研究中应至少包括2个批次。批次数量和放置过的样品的考虑因素应基于使用中稳定性研究所述的一般原则(参见第11.2.1节-批次选择)。此外,申请人可以证明替代策略的合理性,如建模(参见附录2-稳定性建模),以支持短期贮存条件。
The applicant should demonstrate that drug product with a proposed short-term storage condition will remain within the shelf life specifications.
申请人应证明具有建议的短期贮存条件的制剂将保持在有效期质量标准范围内。
11 IN-USE STABILITY
11 使用中稳定性
11.1 Purpose of In-Use Stability Testing
11.1 使用中稳定性试验的目的
This section describes the principles for in-use stability testing for the purpose of establishing or confirming an in-use period and storage conditions, during which the quality of the drug product is maintained within the pre-defined acceptance criteria. In-use conditions are defined as the conditions that mimic the intended use of the drug product after the primary container is first breached and, where applicable, through preparation, storage and administration as per the relevant instructions. The principles outlined in this section are generally applied to single-dose drug products that are handled or prepared and stored prior to administration, including dilution, reconstitution or co-mixing, as well as single containers or combinations of a drug product with a medical device containing drug product intended for multiple administrations or doses. Products packaged in single-use containers for immediate use and not requiring preparation generally do not require an in-use period and would not be subject to in-use stability testing. Assembly of a combination of a drug product with a medical device for immediate use does not constitute preparation in the context of in-use stability testing.
本节描述了使用中稳定性试验的原则,目的是建立或确认使用期间允许时限和贮存条件,在此期间制剂的质量保持在预定的可接受标准范围内。使用条件定义为模拟制剂在主容器首次开启后的预期用途,并在适用的情况下,按照相关说明进行制备、贮存和给药的条件。本节概述的原则通常适用于在给药前处理或制备和贮存的单剂量制剂,包括稀释、复溶或混合,以及单一容器或药械组合,该药械组合包含预期用于多次给药或多剂量的制剂。包装在一次性容器中可立即使用且不需要制备的制剂,通常不需要使用期间允许时限,也不需要进行使用中稳定性试验。在使用中稳定性试验的背景下,药品与医疗器械组合后立即使用不认为是制剂制备。
For a drug product that may remain in contact with a delivery device during administration over time under conditions that differ from the proposed storage (e.g., implantable infusion pump containing the drug product), an in-use study should demonstrate that the drug product remains stable and does not negatively impact the device delivering the drug during the in-use duration.
当制剂在给药期间可能在不同于拟定贮藏条件下与给药装置接触给药(例如,含制剂的植入式输液泵),使用中研究应证明制剂在使用期间保持稳定,且不会对给药装置产生负面影响。
The conditions of use for those products requiring preparation and for multi-dose products may pose a risk to quality of the drug product regarding physicochemical properties and/or microbiological contamination. The regulatory submission for these products should include in-use stability data, upon which the in-use period and instructions are based. This section defines a core framework for establishing or confirming an in-use period and storage conditions, including selection of batches, study design, analytical procedures and acceptance criteria, that are applicable across multiple product types.It is expected that the material in contact with the product and used in the preparation and administration should be demonstrated to be compatible for use with the drug product.
需要制备的制剂和多剂量制剂的使用条件可能会对制剂的理化性质和/或微生物污染质量构成风险。这些制剂的监管递交资料应包括使用中稳定性数据,使用期间允许时限和说明应基于这些数据。本节定义了建立或确认使用期限和贮藏条件的核心框架,包括适用于多种制剂类型的批次的选择、研究设计、分析方法和可接受标准。应证明与制剂接触并用于制备和给药的材料与制剂相容。
Under some circumstances these studies may need to be repeated if certain post-approval variations and changes are made to the product (e.g., formulation, container closure system). To determine whether these studies should be repeated, an assessment of change should be performed according to Section 15.2 - Risk Assessments and Confirmatory Studies to Support Post-Approval Changes.
在某些情况下,如果对制剂(例如,剂型、包装容器和密封系统)进行了某些批准后变更和更改,则可能需要重复这些研究。为确定是否需要重复这些研究,应根据第15.2节-支持批准后变更的风险评估和确认研究进行变更评估。
11.2 In-Use Stability Study Protocol Design
11.2 使用中稳定性研究方案设计
The design of in-use stability study protocols should follow the general principles outlined in Section 3 - Stability Protocol Design. The protocol should simulate the intended use of the product, as detailed in the relevant instructions (e.g., for a multi-dose product stored in a vial, the in-use studies should demonstrate that the container closure system can withstand the conditions of repeated insertion and withdrawal). When designing in-use studies, conditions under which a drug product could be used, including the maximum time the drug product will be exposed to different environmental factors during use, should be considered. For samples requiring preparation, including reconstitution, dilution, or co- mixing, the in-use studies should demonstrate the stability of the product through preparation and handling under the specified storage conditions for the maximum storage period. The study duration, conditions and selection of the analytical procedures and acceptance criteria should be justified as suitable for demonstrating that product quality is maintained throughout the in‐use period. Storage conditions and withdrawal frequency should, at minimum, reflect the instructions-for-use or may consider a worst-case scenario.
使用中稳定性研究方案的设计应遵循第3节-稳定性方案设计中概述的一般原则。方案应模拟制剂的预期用途,详见相关说明(例如,对于贮存在西林瓶中的多剂量制剂,使用期间研究应证明包装容器和密封系统能够承受反复插入和提取的条件)。在设计使用期间研究时,应考虑制剂的使用条件,包括制剂在使用过程中暴露于不同环境因素的最长时间。对于需要制备(包括复溶、稀释或混合)的样品,使用期间研究应证明制剂在规定贮存条件下通过制备和处理达到最长贮存期的稳定性。应证明研究持续时间、条件和分析方法的选择以及可接受标准适用于证明制剂质量在整个使用期间保持不变。贮存条件和提取频率应至少反映使用说明,或可考虑最差的情况。
Alternative (e.g., worst-case) approaches to protocol design may be considered when appropriately justified. For example, for solid oral doses, the applicant may justify the use of open dish studies instead of an in-use study.
适当证明合理时,可考虑方案设计的替代(例如,最差的情况)方法。例如,对于固体口服剂量,申请人可以证明使用开皿研究而不是使用期间研究的合理性。
11.2.1 Selection of Batches
11.2.1 批次选择
Generally, in-use stability data should be provided on two batches of representative drug product. Based on a risk assessment considering product knowledge and available primary stability data, alternative approaches to batch selection may be considered when appropriately justified. At least one of the batches should be chosen towards the end of its shelf life. If such results are not available, one batch should be tested at the final point of the submitted stability studies. If aged batch data are not available at time of filing, a commitment to provide the data or a justification why those data may not be required based on a risk assessment should be provided in the regulatory submission.
通常,应提供两批次代表性制剂的使用中稳定性数据。考虑到产品知识和可用注册批稳定性的数据,基于风险评估,在适当证明合理时,可考虑批次选择的替代方案。至少应选择一个有效期即将结束的批次。如果这些结果不适用,则应在提交的稳定性研究的最终时间点对一批次进行检测。如果在申报时没有适用的老化批次数据,应在监管递交资料中提供提交这些数据的承诺书,或基于风险评估提供为何可能不需要这些数据的理由。
All in-use stability batches should be provided in the container closure system proposed for commercial use (e.g., multi-dose vial, assembled multi-dose combination of a drug product with a medical device), or the administration set up. For drug products presented with different fill volumes, strengths, or presentations, a representative, worst-case or, bracketing or matrixing approach may be applied with justification (refer to Annex 1 – Reduced Stability Protocol Design).
所有使用中稳定性批次应在拟用于商业用途的对于需要制(例如,多剂量小瓶、药械组合的多剂量组合)或给药装置中。对于具有不同装量、规格或剂型的制剂,可采用有代表性的、最差的情况或括号法或矩阵法,并说明理由(参见附录1-简化稳定性方案设计)。
11.2.2 Selection of Analytical Procedures and Acceptance Criteria
11.2.2 分析方法和可接受标准的选择
The analytical procedures with acceptance criteria included in the study should be justified using a risk-based approach that considers the CQAs most likely to change during the proposed in-use period (refer to Section 3.3 - Stability-Indicating Critical Quality Attributes).
应采用基于风险的方法证明本研究中包含的分析方法及其可接受标准的合理性,该方法应考虑在建议的使用期间最有可能发生变更的CQA(参见第3.3节-稳定性指示关键质量属性)。
The analytical procedures should be suitable for the intended purpose and selected to demonstrate the physical, chemical and microbial stability of the product through the proposed in-use period.
分析方法应适用于预期目的,并选择用于证明制剂在建议的使用期限内的物理、化学和微生物稳定性。
For synthetic chemical entities, the physical and chemical quality attributes selected should be appropriate to the individual dosage form and formulation. For example, attributes such as colour, odour,clarity, closure integrity, particulate matter, particle size, moisture content, drug substance assay(s),degradation product level(s), dissolution, antimicrobial preservative and antioxidant content(s), pH and viscosity, and microbial testing should be considered for testing, as applicable with additional considerations for risk associated with dosage form.
对于化学药物,选择的物理和化学质量属性应适合单个剂型和处方。例如,测试时应考虑颜色、气味、澄清度、密封完整性、不溶性微粒、粒度、含水量、原料药含量、降解产物水平、溶出度、抗菌防腐剂和抗氧化剂含量、pH值和粘度以及微生物检测等属性,并酌情考虑与剂型相关的风险。
For biologicals, the physical and chemical quality attributes selected should be appropriate to the individual dosage forms (18). For example, physical and chemical quality attributes of protein content,appearance, clarity, colour, visible particles and high molecular weight species should be tested, unless otherwise justified, while product-related variants or impurities and sub-visible particles should be tested where applicable. Potency testing, or an analytical procedure covering the mode of action, should be included where applicable and potential analytical limitations should be understood. Microbial stability should be assessed through the proposed in-use period for biologicals. Common recommended testing includes a Preservative Efficacy Test (PET) / or Antimicrobial Effectiveness Test (AET), or a microbial enumeration method (e.g., bioburden). In lower risk situations, it may be possible to justify the absence of microbial testing where appropriately justified and based on an assessment of risk.
对于生物制品,选择的物理和化学质量属性应适合单个剂型(18)。 如蛋白质含量、外观、澄清度、颜色、可见异物和高分子量物质等理化质量属性,除非另有说明,在适用的情况下应检测制剂相关变体或杂质和不溶性微粒。在适用的情况下,应包括效价测试或涵盖作用模式的分析方法,并应了解潜在的分析局限性。微生物稳定性应通过生物制品的建议的使用期限进行评估。常见的推荐检测包括防腐剂有效性测试(PET)/或抗菌有效性测试(AET)或微生物计数方法(如生物负荷)。在风险较低的情况下,如果有适当的理由并基于风险评估,可以证明不进行微生物检测是合理的。
11.3 Labelling of the in-use period and storage conditions
11.3 使用期间允许时限和贮存条件的标签
In-use stability data should be used to determine whether a declaration of an in-use period and storage condition are necessary. The in-use period and storage conditions should be stated on the labelling in accordance with regional regulations.
使用中稳定性数据应用于确定是否需要声明使用期间允许时限和贮存条件。应根据地区法规在标签上注明使用期间允许时限和贮藏条件。
There may be scenarios where an established in-use period may not be needed in the labelling. For example, prepared orally administered products, stored in multi-dose containers with a defined supply that is intended for continuous use (not intermittent dosing), may not need to include an in-use period on the labelling if the demonstrated in-use stability data support storage for the intended use of the product.
在某些情况下,标签中可能不需要确定的使用期间允许时限。例如,如果已证明的使用中稳定性数据支持贮存制剂用于预期用途,则可能不需要在标签上注明制备的口服制剂(贮存于多剂量容器中,有固定供应量,供连续使用(非间歇给药))的使用期间允许时限。
12 REFERENCE MATERIALS, NOVEL EXCIPIENTS AND ADJUVANTS
12 参比物质、新型辅料和佐剂
This section covers stability considerations for reference materials, novel excipients (e.g., those used for the first time in a drug product or through a new route of administration) and adjuvants. Novel excipients and adjuvants are discussed due to their significant potential impact on the quality of the drug product.
本节涵盖了参比物质、新型辅料(例如,首次在制剂中使用或通过新给药途径使用的辅料)和佐剂的稳定性考虑。由于新型辅料和佐剂对制剂质量具有显著的潜在影响,因此对其进行了讨论。
Additives (e.g., stabilisers and preservatives) may degrade during the re-test period or shelf life of the drug substance or the shelf life of the drug product. These materials (additives) should be monitored during the stability program if there is an indication that their reaction, degradation, or depletion will adversely affect the quality of the drug product. Refer to Section 3.3 Stability-Indicating Critical Quality Attributes for general stability study design considerations.
添加剂(如稳定剂和防腐剂)可能在原料药的复检期或有效期或制剂的有效期内降解。如果有迹象表明这些材料(添加剂)的反应、降解或消耗会对制剂的质量产生不利影响,则应在稳定性计划期间对这些材料(添加剂)进行监测。有关稳定性研究设计的一般注意事项,请参见第3.3节指示稳定性的关键质量属性。
12.1 Reference Materials
12.1 参比物质
Reference materials (as defined in ICH Q2/Q14), that are used to control the quality attributes of a stored intermediate, drug substance, or drug product should be sufficiently homogenous and stable to ensure scientifically valid results are achieved. If the formulation, material composition, storage condition and/or container closure system for the reference material is different from the drug substance or drug product, a specific reference material stability program may be needed, with an established use period that reflects the differences. Externally sourced, well-characterised reference materials should follow manufacturer recommendations for stability and storage and should be managed within the quality management system (e.g., pharmacopeial materials). Stability data should be available to
support the use period of the in-house reference material. These data are generally provided with the regulatory submission for biologicals and managed within the pharmaceutical quality system (PQS) for synthetics.
用于控制贮存的中间产品、原料药或制剂质量属性的参比物质(定义见ICH Q2/Q14)应具有足够的均匀性和稳定性,以确保获得科学有效的结果。如果参比物质的剂型、材料组成、贮存条件和/或包装容器和密封系统与原料药或制剂不同,则可能需要特定的标准物质稳定性计划,并确定反映差异的使用期限。外部来源的、表征良好的参比物质应遵循生产商关于稳定性和贮存的建议,并应在质量管理体系(如药典材料)内进行管理。应提供稳定性数据以支持内部参比物质的使用期限。这些数据通常与生物制品的监管递交资料一起提供,并在化学药物的药品质量体系(PQS)中进行管理。
12.1.1 Considerations for Synthetic Chemical Reference Materials
12.1.1 化学药物参比物质的注意事项
The use period of a synthetic chemical drug substance, intermediate and drug product reference material may be extended through acceptable stability data and requalification according to established control strategy under a PQS. A synthetic reference material may be stored under more conservative storage conditions than the drug substance and drug product.
化学原料药、中间产品和制剂参比物质的使用期限可通过可接受的稳定性数据并根据PQS下既定的控制策略进行再确认来延长。与原料药和制剂相比,化学药物参比物质可在更保守的贮藏条件下贮存。
12.1.2 Considerations for Biological Reference Materials
12.1.2 生物制品参比物质的注意事项
The use period of a biological reference material, when kept under conditions used to store the corresponding drug substance, intermediate or drug product, should generally be supported by available long-term stability data. When a well-characterised drug substance or drug product is used as an in- house reference material and the storage conditions are the same as that used to store the drug substance or drug product, the drug substance or drug product stability data may support the reference materialuse period, without a need for additional reference material specific stability testing.
生物制品参比物质在用于贮藏相应原液、中间产品或制剂的条件下保存时,其使用期限通常应有长期稳定性数据支持。当使用表征良好的原液或制剂作为内部参比物质,并且贮藏条件与用于贮藏原液或制剂的条件相同时,原液或制剂的稳定性数据可以支持标准物质的使用期限,而不需要额外的标准物质特定稳定性试验。
Alternative storage conditions may extend the use period of in-house biological reference materials beyond the re-test period or shelf life of the drug substance, intermediate, or drug product (e.g.,stabilising storage at a sufficiently lower temperature than the drug substance or drug product storage condition). The alternative storage condition should be justified with its own long-term stability data or a concurrent stability testing strategy that allows for a trend analysis of the data. The reference material use period may be extended through acceptable stability data according to a protocol (e.g., qualification).
其他贮藏条件可能会延长内部生物制品标准物质的使用期限,使其超过原液、中间产品或制剂的复检期或有效期(例如,在比原液或制剂贮藏条件低得多的温度下稳定贮藏)。其他贮藏条件应通过其自身的长期稳定性数据或允许对数据进行趋势分析的同步稳定性试验策略来证明。根据方案(例如,确认),可通过可接受的稳定性数据延长参比物质的使用期限。
In situations where a drug substance or product’s stability-indicating critical quality attribute (e.g.,potency) is being controlled relative to a reference material, a risk-based approach, including more stringent stability acceptance criteria and trend analyses, should be considered for the reference material’s stability to prevent drift in the stability profile of the drug substance or product.
在原液或制剂的稳定性指示关键质量属性(如效价)相对于参比物质受到控制的情况下,应考虑对参比物质的稳定性采用基于风险的方法,包括更严格的稳定性可接受标准和趋势分析,以防止原液或制剂的稳定性特征发生漂移。
12.2 Novel Excipients
12.2 新型辅料
Novel excipients should be evaluated for their impact on the stability of the drug product and relevant information should be included in the regulatory submission following the recommendations described in the applicable sections within this guideline (refer to Section 3 - Stability Protocol Design, Section 6-Testing Frequency and Section 7 - Storage Conditions).
应评价新型辅料对制剂稳定性的影响,并按照本指导原则适用章节中所述的建议(参见第3章-稳定性方案设计、第6章-试验频率和第7章-贮藏条件),将相关信息纳入监管递交资料。
If the excipient itself is a protein (e.g., albumin) and used with a biological drug substance, additional risk assessments should be provided to clarify the known degradation profile of the excipient and its impact on the biological drug substance or drug product. For protein-based excipients, the drug product stability studies should address their potential protein-excipient interaction, quantity of intact excipient in the drug product and impact on drug product immunogenicity as well as their potential for masking process related impurities.
如果辅料本身是蛋白质(如白蛋白),并与生物制品原液一起使用,则应提供额外的风险评估以阐明辅料的已知降解产物谱及其对生物制品原液或制剂的影响。对基于蛋白质的辅料,制剂稳定性研究应说明其潜在的蛋白质-辅料相互作用、制剂中完整辅料的数量和对制剂免疫原性的影响,以及掩盖工艺相关杂质的可能性。
12.3 Vaccine Adjuvants
12.3 疫苗佐剂
Adjuvant stability data should be provided in the regulatory submissions for vaccines. Stability of the adjuvant should be assessed by formal stability studies. If alternative strategies for determining stability of the adjuvant are potentially applicable, the applicant should consider early engagement with the regulatory authority.
疫苗的监管递交资料中应提供佐剂稳定性数据。佐剂的稳定性应通过正式稳定性研究进行评估。如果确定佐剂稳定性的替代策略可能适用,申请人应考虑尽早与监管机构联系。
The stability studies will depend on the formulation/presentation, where vaccine drug product formulated with the adjuvant will have different consideration to formulations where the adjuvant is provided in a separate vial to the vaccine drug product. For adjuvants that are mixed with the drug substance at the production site to derive the adjuvanted vaccine drug product, data that support shelf life of the adjuvanted vaccine in the primary container is required. In case of adjuvanted vaccines that depend on antigen adsorption to the adjuvant (e.g., alum/antigen mixture) stability monitoring should consider the degree of antigen adsorption/binding and extent of dissociation of antigen from the adjuvant upon storage, where relevant.
稳定性研究将取决于剂型/规格,其中用佐剂配制的疫苗制剂与单独装瓶的疫苗制剂有不同的考虑因素。对于在生产场地与原液混合以获得含佐剂疫苗制剂的佐剂,需要支持主容器中佐剂疫苗有效期的数据。如果依赖于抗原吸附到佐剂上的佐剂疫苗(如明矾/抗原混合物),则稳定性监测应考虑抗原的吸附/结合程度以及贮藏后抗原与佐剂的解离程度。
When the adjuvant and vaccine antigen (vaccine components) are supplied in separate containers, the stability of each component should be assessed following appropriate pre-defined protocols that reflect storage duration and storage conditions of each vaccine component.
当佐剂和疫苗抗原(疫苗组分)以单独的容器供应时,应按照适当的预定义方案评估每种组分的稳定性,这些方案反映每种疫苗组分的贮藏期限和贮藏条件。
The in-use stability of the adjuvant-antigen mixture should be assessed in the situation when the mixture is not administered immediately after preparation and should be performed at the intended in-use conditions and period (refer to Section 11 – In-Use Stability). It is important to set appropriate acceptance criteria to assess integrity of the adjuvant in the adjuvant/vaccine antigen mixture. The data generated in the in-use stability studies will support the instructions for use of the admixed vaccine.
佐剂-抗原混合物的使用中稳定性应在混合液制备后不立即给药的情况下进行评估,并应在预期的使用条件下和期限内进行(参见第11节-使用中稳定性)。制定适当的可接受标准以评估佐剂/疫苗抗原混合物中佐剂的完整性非常重要。使用中稳定性研究生成的数据将支持混合疫苗的使用说明。
13 DATA EVALUATION
13 数据评价
13.1 General Considerations
13.1 一般考虑因素
Stability data are obtained for multiple purposes throughout the product lifecycle. A systematic approach should be adopted in the presentation and evaluation of the stability information. This section focuses on the evaluation of stability data to establish a re-test period or shelf life for drug substance and the shelf life for drug product based on long-term data at the recommended storage condition. Refer to Section 3 - Stability Protocol Design, Table 1 for the minimum stability data at the time of submission.
在整个产品生命周期中,稳定性数据可用于多种目的。稳定性资料的陈述和评价应采用系统的方法。本节重点对稳定性数据进行评价,以根据推荐贮藏条件下的长期数据确定原料药的复检期或有效期以及制剂的有效期。递交时的最低稳定性数据见第3节-稳定性方案设计,表1。
Alternatively, when there is limited long-term stability data at the recommended storage condition, the re-test period or shelf life can be proposed based on:
或者,当在推荐的贮藏条件下长期稳定性数据有限时,可基于以下因素拟定复检期或有效期:
• Use of enhanced stability modelling methodologies to predict or extrapolate the stability profile past the point of the available real-time data (refer to Annex 2 – Section A2-2- Enhanced Stability Modelling).
• 使用增强的稳定性建模方法来预测或外推超过可用实时数据点的稳定性特征(参见
附录2第A2-2节-增强的稳定性建模)。
• Limited extrapolation of the real-time data for synthetic chemical entities that may be supported by accelerated condition stability data using a decision tree approach. For biologicals, the decision tree approach, which is based on the extent of attribute change at accelerated storage conditions, is not considered suitable due to the inherent differences in degradation mechanisms and other structure/function differences within biologicals.
• 化学合成实体实时数据的有限外推,可以由使用决策树方法的加速条件稳定性数据支持。对于生物制品,由于生物制品内固有的降解机制差异和其他结构功能差异,基于加速条件下属性变化程度的决策树方法不适用。
A comprehensive stability data evaluation should take into consideration any stored intermediates, process hold times, any short-term storage outside of the long-term storage conditions, including the risk of excursions to the storage conditions and manipulations of the product to the completion of administration to the patient (in-use stability).
全面的稳定性数据评价应考虑任何贮藏的中间产品、工艺保持时间、长期条件之外的任何短期贮藏,包括偏离贮藏条件的风险和完成患者给药前的制剂操作(使用中稳定性)。
Guidance is provided for the data evaluation of drug substance and drug product that have stability data from at least three primary batches with batch as a single factor, and multi-factor products with full design (for example, products with the same drug substance at different fill volumes, varied concentration, container closure system dimensions, etc.). In addition, the degree of variability between batches and other factors affect the confidence that a future production batch will remain within acceptance criteria throughout its re-test period or shelf life. Multi-factor products with reduced design studies are discussed in Annex 1 - Reduced Stability Protocol Design.
为具有至少三个注册稳定性批次稳定性数据且批次为单因素的原料药和制剂,以及具有完整设计的多因素制剂(例如相同原料药不同装量、不同浓度、包装容器和密封系统等)的数据评价提供指导。此外,批次之间的变异程度和其他因素将影响以后的产品批次在整个复检期或有效期中能符合质量标准的可靠程度。简化设计研究的多因素制剂在附录1-简化稳定性方案设计中进行了讨论。
When the principles for extrapolation and modelling are considered to apply to other product types, such as ATMPs or vaccines, the applicant should seek early engagement with the regulatory authority.
当外推和建模原则被认为适用于其他制剂类型(如ATMP或疫苗)时,申请人应尽早与监管机构联系。
13.1.1 Re-Test Period
13.1.1 复检期
A re-test period is normally applicable to drug substances of synthetic chemical entities as an alternative to establishing a shelf life. This approach may also be proposed in certain cases for the drug substances of biologicals with a well understood stability profile, where justified. An example where a re-test period may apply for a biological drug substance is a well characterised IgG therapeutic monoclonal antibody that is stored frozen and shows little to no change in product quality over the duration of storage.
复检期通常适用于化学合成实体的原料药,作为确定有效期的替代方案。在某些情况下,如果合理,对于具有良好稳定性特征的生物制品原液,也可以提出该方法。生物制品原液可申请复检期的一个例子是,表征良好的IgG治疗性单克隆抗体冷冻贮藏,在贮藏期间制剂质量几乎没有变化。
13.1.2 Start of Shelf Life for Synthetic Chemical Entity Drug Products
13.1.2 化学合成实体制剂有效期起始
The start of shelf life should be the date of production, which is defined as the date of the first manufacturing step that combines drug substance with other ingredients.
In accordance with regional requirements, consider the following approaches:
有效期的起始日期应为生产日期,即原料药与其他成份混合的第一个生产步骤的日期。
根据区域要求,考虑以下方法:
• When the date of release is less than 30 days from the date of production, the start of shelf life of a drug product batch could instead be calculated from the date of release of that batch.
• 当放行日期距生产日期少于30天时,制剂批次的有效期起始日期可以从该批次的放
行日期开始计算。
• For drug products consisting of a drug substance as a single ingredient, filled into the final drug product container, the initial date of the filling operation is taken as the date of production.
• 对于将原料药作为单一成份灌装至最终制剂容器的制剂,将灌装操作的初始日期视为生产日期。
In the case of a drug product intermediate storage step before further processing and when the start of shelf life is not defined as described above, these should be declared and justified and included in the drug product stability program of batches that represent the cumulative maximum holding times of drug product intermediates.
对于进一步加工前的制剂中间产品贮存步骤,如果未如上所述定义有效期的起点,则应声明并证明其合理性,并将其纳入代表制剂中间产品累积最大放置时间的批次的制剂稳定性计划中。
13.1.3 Start of Shelf Life for Biological Drug Products
13.1.3 生物制剂有效期起始
The start of shelf life for biological drug products begin on the date of manufacture e.g., date of filtration and/or filling for a liquid drug product. When the drug product filling operation takes place over more than one day, then the initial date of the filling operation is taken as the date of manufacture. Other approaches used to define the start of shelf life can be used if justified.
生物制剂的有效期从生产日期开始,例如液体制剂的过滤和/或灌装日期。当制剂灌装操作超过一天时,则将灌装操作的初始日期作为生产日期。在合理的情况下,可以使用其他方法定义有效期的起始时间。
13.2 Statistical Evaluation of the Long-term Storage Condition Stability Profile to Establish the Re-test Period or Shelf Life
13.2 确定复检期或有效期的长期条件稳定性特征的统计评价
All stability data from the primary and supportive stability studies should be evaluated to establish a re-test period or shelf life. The statistical evaluation should include all primary stability studies, any available production scale studies and supplemented, when applicable, with additional supportive data from batches included in the stability programme (refer to Section 4 - Selection of Batches). The stability profiles for the CQAs shown to potentially change over time at the recommended storage conditions should be evaluated to establish the re-test period or shelf life. Each CQA should be assessed separately, and an overall assessment should be made of findings for the purposes of proposing a shelf life or re-test period. The re-test period or shelf life proposed should not exceed that predicted for anysingle attribute.
应对注册和支持性稳定性研究的所有稳定性数据进行评价,以确定复检期或有效期。统计学评价应包括所有注册稳定性研究、任何可用的生产规模研究,并在适用时补充稳定性计划中批次的其他支持性数据(参见第4章-批次选择)。对于在推荐的贮藏条件下可能随时间变化的CQA,应评价其稳定性特征,以确定复检期或有效期。应单独评估每个CQA,并对结果进行总体评估,以确定有效期或复检期。所建立的复检期或有效期不能超过单个质量指标预示的有效期限。
Data from quantitative analytical procedures should be evaluated using appropriate statistical tools;whereas results from semi-quantitative or qualitative analytical procedures, which may not be amenable to statistical analysis, should also be evaluated. The degree of variability across individual batches and the number of data time-points affects the confidence that a future production batch will remain within specification throughout the established re-test period or shelf life (24).
应使用适当的统计工具对定量分析程序的数据进行评价;而半定量或定性分析程序的结果可能不适合进行统计分析,因此也应对其进行评价。批次之间的变异程度和数据时间点的数量将影响以后的产品批次在整个确定的复检期或有效期中能符合质量标准的可靠程度(24)。
There are many valid statistical methods to evaluate stability data to set a re-test period or shelf life from batches of substances, intermediates, or products. The statistical methodology used should be justified as suitable for the product type, the data set used for the analysis (batches, study design factors, etc.) and the purpose of the evaluation. The following sections outline selected, commonly used approaches and do not cover all situations (26, 27).
有许多有效的统计方法来评估稳定性数据,以设定各批次原料药、中间产品或制剂的复检期或有效期。应证明所使用的统计方法适用于产品类型、用于分析的数据集(批次、研究设计因素等)和评价目的。以下各节概述了选定的常用方法,但并未涵盖所有情况(26、27)。
13.2.1 Linear Regression for an Individual Batch
13.2.1 单一批次的线性回归
Each primary batch, stored under the long-term conditions, may be evaluated individually to establish the re-test period or shelf life. Where there are differences in stability observed among batches or among other factors or factor combinations that preclude the combining of data, the proposed re-test period or shelf life should not exceed the earliest time (worst-case) period supported by any batch, other factor, or factor combination. For quantitative attributes expected to change with time following a linear pattern or log transformed data that follow a linear pattern at the recommended storage condition, an approach for evaluating the data is by linear regression analysis. The appropriateness of the assumed linear relationship over time and normal distribution of the variables may be supported by evaluation of the residuals for the regression line (goodness of fit).
在长期条件下贮藏的每个注册稳定性批次可单独评价,以确定复检期或有效期。如果不同批次间或其他因素或因素组合间观察到的稳定性差异导致数据无法合并,则建议的复检期或有效期不应超过由批次、其他因子或因子组合中所支持的最短时间(最坏情况)期限。对于在推荐的贮藏条件下遵循线性模式且预计随时间变化的定量指标或遵循线性模式的对数转换数据,可通过线性回归分析评估数据。假设的随时间变化的线性关系和变量正态分布的适当性可以通过评价回归线的残差(拟合度)来支持。
Analyses of a quantitative attribute can be performed by determining the earliest time at which the 95% percent confidence limit for the mean intersects the proposed acceptance criterion. For attributes with upper and lower acceptance criteria, a two-sided 95% confidence limit is recommended. The point at which the confidence limits for the mean intersects the acceptance limit for each individual batch under evaluation is generally determined (illustrated in Annex-2 Stability Modelling for an individual batch example). Using this approach, the upper and lower limits may each be evaluated individually as one-sided limits against their respective upper and lower acceptance criteria. For attributes with only a lower or an upper acceptance criterion, such as those for purity/impurity, a one-sided 95% confidence limit is recommended.
某一定量指标分析可以通过确定平均值的95%置信限与建议的认可标准(限度)相交的第一时间点来定。对于认可标准具有上限和下限的质标,建议使用双侧95%置信限。通常确定平均值置信限与接受评价的每个单一批次的可接受限值相交的点(如附录2单一批次示例的稳定性建模所示)。使用这种方法,上限和下限可以分别作为单侧限值根据各自的可接受标准的上限和下限进行评估。当指标仅有认可标准的上限或下限,如纯度/杂质,推荐使用单侧95%置信限。
Re-test period or shelf life for individual batches should first be estimated with individual intercepts,individual slopes and the pooled mean square error calculated from all batches. If each batch has an estimated re-test period or shelf life longer than that proposed, the proposed re-test period or shelf life will generally be considered appropriate. If, however, one or more of the estimated re-test periods or shelf lives are shorter than that proposed, a statistical test can be performed to determine whether the batches can be combined to estimate a longer re-test period or shelf life.
首先采用各个批次的截矩、各个斜率和从全部批次计算得平均方差,估算各个批次的复检期或有效期。如果估算的每批的复检期或有效期比建议的长,一般认为建议的复检期或有效期是合适的。但是,如果一个或多个估算的复检期或有效期比建议的短,应进行统计检验,以确定能否合并这些批次以评估一个比较长的复检期或有效期。
13.2.2 Combining Batches
13.2.2 合并批次
For the statistical evaluation, it may be advantageous to combine the data from different representative batches into one overall estimate. A linear regression analysis provides a test for the parameters that define the linear stability profile of an attribute from a single batch and whether they can be combined to determine: first the change over time or slope followed by the y-intercept. An appropriate statistical approach should be prospectively defined and justified to evaluate the ability of combining data from different batches (22, 23). Refer to Annex 2 - Stability Modelling for additional statistical considerations.A simulation study can be useful, if applicable, to demonstrate that the statistical properties of the procedure selected are appropriate (25).
对于统计评价,将不同代表性批次的数据合并为一个总体估计值可能是有利的。线性回归分析用于检测定义单个批次属性的线性稳定性特征的参数,以及是否可以合并这些参数来确定:首先是随时间或斜率的变化,然后是y轴截距。应预先定义并证明适当的统计方法,以评估合并不同批次数据的能力(22,23)。有关其他统计考虑因素,请参见附录2-稳定性建模。如可能,可使用模拟研究来证明所选方法的统计性质是合适的(25)。
13.2.3 Scale Transformation of Data
13.2.3 数据规模转换
When the degradation kinetics are complex and decelerating (e.g., a biphasic degradation profile characterised by fast initial rate followed by a slower longer-term rate or when the data that may show a plateauing profile), a linear regression analysis could be proposed when the linear regression provides a worst-case shelf life or re-test period. The nature of the relationship between an attribute and time will determine whether data should be transformed for linear regression analysis. The relationship can be represented by a linear or non-linear function on an arithmetic or logarithmic scale. In some cases, a non-linear regression can better reflect the true relationship. It should be noted that in some instances if a linear function is fit to plateauing data, data points beyond the plateau could skew the regression line towards later timepoints. Whereas this section describes linear regression analysis, other approaches may be used (e.g., nonlinear regression) with justification. When scale of transformation is used, statistical methods should be prospectively employed to evaluate the goodness of fit on all batches and combined batches (where appropriate) to the inferred degradation profile. Transformation of a non-linear model should be justified from a scientific perspective (e.g., understanding of the attribute and/or analytical procedure).
当降解动力学复杂且缓慢时(例如,双相降解产物谱的特点是初始速率快,随后为较慢的长期速率,或者这时的数据可能显示出平台期),当线性回归提供最坏情况下的有效期或复检期时,可建议进行线性回归分析。定量指标与时间之间的关系决定了这些数据是否需进行转换以进行线性回归分析。一般这种关系可用算术或对数坐标中的线性或非线性函数来表示。有时非线性回归能更好反映其真实关系。应该注意的是,在某些情况下,如果线性函数拟合平台期数据,平台期之外的数据点可能会使回归线向较晚的时间点倾斜。虽然本节描述了线性回归分析,但也可以使用其他方法(例如非线性回归),但需证明其合理性。当使用规模转换时,应预先采用统计方法来评价所有批次和组合批次(如适用)与推断降解产物谱的拟合度。应从科学角度(例如,对属性和/或分析方法的理解)证明非线性模型转换的合理性。
13.2.4 Extrapolation and Stability Modelling
13.2.4 外推和稳定性建模
Extrapolation is the practice of using a known data set to infer information about future data and is a form of stability modelling that, under certain conditions, may be applicable to synthetics and biologicals. Extension of shelf life beyond the period covered by long-term data, by extrapolation, can be proposed in the regulatory submission. Whether extrapolation of stability data is appropriate depends on the extent of understanding for the product type, relevant knowledge about the stability-indicating attributes and any change over time, the goodness of fit of any mathematical or other computational model type, and the existence of relevant supporting data that may include additional timepoints, additional batches or prior knowledge. Relevant supporting data include satisfactory long-term data from development batches that are (1) made with a comparable formulation to, (2) manufactured on a smaller scale than, or (3) packaged in a container closure system similar to that of the primary stability batches.
外推法是一种根据已知数据来推断未来的数据的方法,也是一种稳定性建模形式,在某些条件下可能适用于合成药物和生物制品。在监管提交中,可用外推法建立超过长期试验数据覆盖时间范围的有效期。稳定性数据外推是否适用取决于对产品类型的了解程度、关于稳定性指示属性和随时间变化的相关知识、任何数学或其他计算模型类型的拟合度,以及是否存在相关支持性数据,可能包括额外时间点、额外批次或先验知识。相关支持性数据包括来自开发批次的符合要求的长期数据,这些开发批次:(1)采用与注册稳定性批次具有可比性的处方,(2)以较小的规模生产,或(3)包装在与注册稳定性批次相似的容器密封系统中。
For synthetics, certain quantitative chemical attributes (e.g., assay, chemical degradation products, preservative content) for a drug substance or product can generally be assumed to follow zero-order kinetics during long-term storage. Although the kinetics of other quantitative attributes (e.g., pH, dissolution) are generally not known, the same statistical analysis can be applied, if appropriate.Qualitative attributes and microbiological attributes are not amenable to this kind of statistical analysis.The decision tree approach would not be recommended for biological products because biological and immunological attributes are generally not amenable to extrapolation, as they cannot be assumed to follow zero order kinetics. For certain well characterised biologicals that have no statistically significant or meaningful change over time, extrapolation may be possible using the risk assessment criteria and supporting long term development data, as outlined in Section 13.2.9 – Extrapolation of Biologicals.
对于化学药物,一般可假定原料药或制剂的某些可定量的化学指标(如:含量,降解产物,防腐剂含量)在长期贮藏期间符合零级反应动力学。尽管其他一些可定量指标(如pH值,溶出度)的动力学尚不明确,如合适,也可用上述的统计分析方法。定性指标和微生物属性通常不适合这一类的统计分析。对于生物制品,不建议使用决策树方法,因为生物和免疫学属性通常不适合外推,因为不能假定它们符合零级动力学。对于某些已充分表征的生物制品,如果随时间推移未发生统计学显著或有意义的变化,则可使用风险评估标准和支持性长期研发数据进行外推,如第13.2.9节“生物制品外推”所述。
An extrapolation of stability data assumes that the same change profile will continue to apply beyond the period covered by available long-term data and should be applicable to future batches. The correctness of the assumed change profile is a critical consideration, especially when stability data are limited. Any extrapolation should be justified and have a science-based rationale that may be based on prior knowledge.
稳定性数据的外推假定相同的变化特征将在现有长期数据覆盖时间段之后继续适用,并应适用于未来批次。当稳定性数据有限时,所假定的变化特征的正确性是关键考虑因素。任何外推都应是合理的,并具有可能基于先验知识的科学依据。
The methodologies outlined in this section may be used to extrapolate the long-term stability data.When estimating a regression line or curve to fit the long-term data, the data themselves provide a check on the correctness of the assumed change pattern, and statistical methods should be applied to evaluate the goodness of fit (or an equivalent valid statistical method) of the existing data to the inferred line or curve and to provide confidence that future batches will lie within the inferred stability profile (refer to Annex 2, Section A2-1 - Statistical Evaluation of Stability Data from Single or Multi-factor Study Designs). No such internal check is possible beyond the period covered by long-term data from primary batches, though an inferred trend may be supported by prior knowledge.
本节中概述的方法可用于外推长期稳定性数据。当判断长期数据是否符合直线回归或曲线回归时,数据本身也可验证所假设的变化模式的正确性,并可用统计方法评估现有数据与推断直线或曲线的拟合度(或等效的有效统计方法),并提供置信度,即未来批次将位于推断的稳定性特征范围内(参见附录2第A2-1节-单因素或多因素研究设计的稳定性数据的统计评估)。尽管推断的趋势可能得到先验知识的支持,在注册稳定性批次长期数据覆盖的时间范围外,不可能进行这种相互验证。
Enhanced stability modelling, such as those referenced in Annex 2 (Annex 2- Section A2-2 Enhanced Stability Modelling) may also be considered.
也可考虑增强的稳定性建模,如附录2(附录2-第A2-2节:增强的稳定性见建模)中引用的建模。
Any shelf life or re-test period proposed based on extrapolation should be verified by additional long-term stability data as these data become available.
因此,由外推法建议的有效期或复检期,应及时采用后续得到的长期稳定性数据进行验证。
13.2.5 Extrapolation for Synthetic Chemical Entities
13.2.5 化学合成实体的外推
A systematic approach using a decision tree (Figure 4) is provided as a tool for appropriate data extrapolation beyond the period covered by long-term stability data. The decision tree is intended to apply to synthetic chemical entities that are stored long-term at room temperature or refrigerated conditions and that have stability data at an accelerated storage condition in addition to the long-term stability data. The decision tree is not intended for other products or other long-term conditions (e.g.,biologicals or frozen storage). The decision tree provides a complementary approach to the statistical analysis of long-term stability data. The decision tree approach may provide some limited extrapolation though greater extrapolation beyond these stated limits may be possible using other modelling methodologies (refer to Annex 2 –Stability Modelling).
提供了一种使用决策树的系统方法(图4),作为在长期稳定性数据覆盖时间范围外进行适当数据外推的工具。决策树适用于在室温或冷藏条件下长期贮藏,且出长期稳定性数据外,还具有加速条件稳定性数据的的化学合成实体。决策树不适用于其他产品或其他长期条件(例如,生物制品或冷冻贮藏)。决策树为长期稳定性数据的统计分析提供了一种补充方法。决策树方法可以提供一些有限的外推,不过使用其他建模方法也可能实现超出这些规定限值的更大外推(参见附录2-稳定性建模)。
To use the decision tree, the variability between and within batches should allow reasonable confidence that the stability profile meets the attribute specification at the proposed re-test period or shelf life under the recommended storage conditions. The term “room temperature” refers to the general customary environment and should not be inferred to be the storage statement for labelling (refer to Section 14 –Labelling).
为了使用决策树,批次间和批次内的变异程度应允许有合理的置信度,即在建议的复检期或有效期内,在推荐的贮藏条件下,稳定性特征符合属性质量标准。术语“室温”指一般环境,不应推断为标签上的贮藏说明(参见第14节-说明书及标签)。
Figure 4: Decision Tree for Data Evaluation for Re-test Period and Shelf Life Estimation for
Synthetic Chemical Entity Drug Substances and Drug Products (excluding frozen
products)
图4 建立化学合成实体原料药或制剂(除冷冻制剂外)复检期和有效期的数据评价决策树
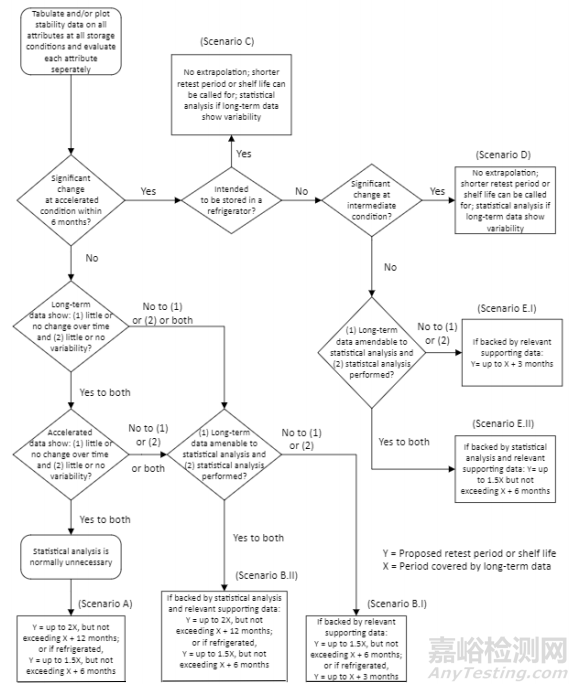
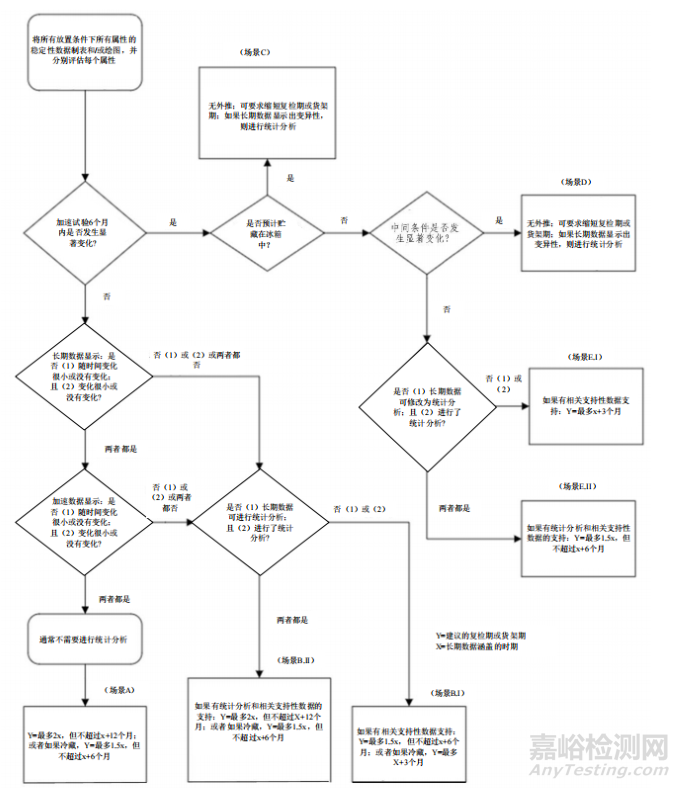
When the decision tree is used for extrapolation, each attribute on the shelf life specification should be systematically evaluated. The assessment should begin with any significant change at the accelerated condition and, if appropriate, at an intermediate condition, and progresses through the trends and variability of the long-term data. The circumstances are delineated under which extrapolation of re-test period or shelf life beyond the period covered by long-term data can be appropriate. If any attribute that is not quantifiable shows potential for significant change at the accelerated storage condition, then the decision tree cannot be used.
The following subsections describe the decision tree approach, and the scenarios illustrated.
当决策树用于外推时,应对有效期质量标准的每个属性进行系统评估。评估应从加速条件下或在中间条件下出现的任何明显变化开始,至整个长期试验结果的变异性和趋势。在某些情况下,可超过长期试验数据所覆盖的时间范围,外推复检期或有效期。如果任何不可量化的属性在加速条件下表现出显著变化的可能性,则不能使用决策树。
以下小节描述了决策树方法和演示场景。
13.2.6 No Significant Change at Accelerated Condition
13.2.6 在加速条件下无明显变化
Where no significant change occurs at the accelerated condition, the re-test period or shelf life would depend on the nature of the long-term and accelerated data. This applies to room temperature and refrigerated drug substances and drug products where no significant change occurs at the accelerated condition.
如果在加速条件下没有发生显著变化,复检期或有效期将取决于长期和加速数据的性质。这适用于贮藏于室温和冷藏条件下、在加速条件下不发生显著变化的原料药和制剂。
13.2.6.1 Long-term and Accelerated Data Show Little to No Change Over Time and Little or No Variability (Scenario A)
13.2.6.1长期和加速数据显示,随时间推移变化很小或没有变化,变异很小或没有变异(场景A)
Where the long-term data and accelerated data for an attribute show little or no change over time and little or no variability, it might be apparent that the drug substance or product will remain well within the acceptance criteria for that attribute during the proposed re-test period or shelf life. In these circumstances, a statistical analysis is normally considered unnecessary but justification for the omission should be provided. Justification can include a discussion of the change pattern or lack of change, relevance of the accelerated data, mass balance, and/or other supporting data. Extrapolation of the re-test period or shelf life beyond the period covered by long-term data can be proposed. The proposed re-test period or shelf life can be up to two times for products stored at room temperature, but should not be more than 12 months beyond, the period covered by long-term data. For refrigerated drug substances or drug products, if the long-term and accelerated data show little change over time and little variability, the proposed re-test period or shelf life can be up to one-and-a-half times, but should not be more than 6 months beyond the period covered by long-term data.
当某一指标的长期数据和加速数据随时间变化很小或几乎没有变化,变异性也很小或几乎没有变异性时,原料药或制剂在建议的复检期或有效期内仍能较好地符合该指标的可接受标准。
在这些情况下,通常认为没有必要做统计分析,但应说明将其省略的具体理由。理由可包括对变化模式或缺乏变化的讨论、加速数据的相关性、质量平衡和/或其他支持性数据。可以建议将复检期或有效期外推至长期数据涵盖的时间周期以外。对于在室温下贮藏的产品,建议的复检期或有效期可长达2倍,但不应超过长期数据涵盖时间后12个月。对于冷藏原料药或制剂,如果长期和加速数据显示其随时间变化不多、变异性不大,建议的复检期或有效期可长达1.5倍,但不应超过长期数据涵盖时间后6个月。
13.2.6.2 Long-term or Accelerated Data Show Change Over Time and/or Variability
(Scenario B)
13.2.6.2长期或加速数据显示随时间变化和/或变异(场景B)
The decision tree approach considers the significance of change over time under accelerated and long-term storage conditions and method variability. For a synthetic chemical drug substance, a significant change is when an attribute exceeds specification at the accelerated condition within 6 months or long-term storage condition within the intended shelf life or re-test period. For drug product, a significant change has additional considerations applicable to synthetic chemical products including:
决策树方法考虑了加速和长期条件下随时间变化的重要性以及方法变异性。对于合成化学原料药,显著性变化是指在6个月内加速条件下或在预期有效期或复检期内长期条件下,某个属性超过质量标准。对于制剂,显著变化有适用于合成化学产品的额外考虑因素,包括:
(1)5% change in assay from its initial value
(1) 含量与初始值相比变化5%
(2) failure to meet the specification for degradation products, physical attributes (e.g., colour, phase separation, re-suspendability, caking, hardness) and, when applicable functionality tests (e.g., dose delivery per actuation);and for certain dosage forms:
(2) 不符合降解产物、物理属性(如:颜色、相分离、再分散性、结块、硬度)以及适用的功能性测试(如每揿剂量)的质量标准;对于某些剂型:
(3) failure to meet specification for pH
(3) pH值不符合质量标准
(4)failure to meet specification for dissolution testing
(4) 不符合溶出度检测质量标准
With respect to physical attribute changes, the following can be expected to occur at the accelerated condition and would not be considered significant change that calls for intermediate testing if there is no other significant change:
关于物理属性变化,在加速条件下将预期发生以下情况,如果没有其他显著变化,则不会被视为需要进行中间试验的显著变化:
softening of a suppository that is designed to melt at 37°C, if the melting point is clearly
demonstrated, failure to meet acceptance criteria for dissolution of a gelatine capsule or gel-coated tablet if the failure can be unequivocally attributed to cross-linking.
设计在37°C融化的栓剂的软化情况,如果熔点得到明确显示,不符合明胶胶囊或凝胶包衣片溶出度的可接受标准,如果明确归因于交联。
However, if phase separation of a semi-solid dosage form occurs at the accelerated condition, testing at an intermediate condition should be performed. Potential interaction effects (e.g., other drug product components) should also be considered in establishing that there is no significant change.
但是,如果半固体剂型在加速条件下发生相分离,则应进行中间条件试验。在确定无显著变化时,还应考虑潜在的交互作用(比如其他制剂组分)。
For product intended to be stored at room temperature, when a significant change is observed or anticipated at a particular accelerated storage condition, consider including an intermediate storage condition in the protocol and for the data evaluation. An appropriate intermediate storage condition, as applied to a synthetic chemical entity, depends on the climatic zones intended for the product (refer to Section 7 – Storage Conditions).
对于拟在室温下贮藏的产品,当在特定的加速条件下观察到或预期会发生显著变化时,应考虑将中间条件纳入方案和数据评估中。适用于化学合成实体的适当中间条件取决于产品的预期气候带(见第7节“贮藏条件”)。
If the long-term or accelerated data for an attribute show change over time and/or variability within a factor or among factors (e.g., strength, container size and/or fill), statistical analysis of the long-term data can be useful in establishing a re-test period or shelf life. When there are differences in stability observed across batches or among other factors or factor combinations (e.g., strength, container size and/or fill) that preclude the combining of data, the proposed re-test period or shelf life should not exceed the shortest period supported by any batch, other factor, or factor combination. Alternatively, where the differences are readily attributed to a particular factor (e.g., strength), different shelf lives can be assigned to different levels within the factor (e.g., different strengths). A discussion should be provided to address the cause for the differences and the overall significance of such differences on the product. Extrapolation beyond the period covered by long-term data can be proposed; however, the extent of extrapolation would depend on whether long-term data for the attribute are amenable to statistical analysis.
如果某一属性的长期或加速试验数据显示随时间的变化和/或在某个因素内或多个因素(如规格、容器大小和/或装量)之间存在变异性,则对长期数据的统计分析可用于确定复检期或有效期。如果在批次之间、或者其他因素或因素组合(如规格、容器大小和/或装量)之间观察到稳定性存在差异而无法合并数据时,建议的复检期或有效期不应超过任何批次、其他因素或因素组合所支持的最短期限。或者,如果差异容易归因于特定因素(如规格),则可以将不同的有效期指定给该因素内的不同水平(如不同规格)。应提供相关讨论,以说明差异的原因以及此类差异对产品的整体重要性。可以建议外推至长期数据涵盖时间周期之外;但外推的范围将取决于该属性的长期数据是否适合进行统计分析。
13.2.6.3 Data not amenable to statistical analysis (Scenario B.I)
13.2.6.3 数据不可用于统计分析(场景B.I)
Where long-term data are not amenable to statistical analysis (e.g., colour, clarity using qualitative or semi-quantitative methods), but change over time and relevant supporting data are provided, theproposed re-test period or shelf life at room temperature storage can be up to one and-a-half times but should not be more than 6 months beyond the period covered by long-term data. For refrigerator storage, the proposed re-test period or shelf life can be up to 3 months beyond the period covered by long-term data.
如果长期数据不适合进行统计分析(比如使用定性或半定量方法的颜色、澄清度),但提供了随时间的变化和相关支持性数据,建议的复检期或室温贮藏的有效期可长达1.5倍,但不应超过长期数据涵盖时间后6个月。对于冷藏保存,建议的复检期或有效期最长可达长期数据涵盖时间后3个月。
13.2.6.4 Data amenable to statistical analysis (Scenario B.II)
13.2.6.4 数据可用于统计分析(场景B.II)
If long-term data are amenable to statistical analysis but no analysis is performed, the extent of extrapolation should be the same as when data are not amenable to statistical analysis. However, if a statistical analysis is performed, it can be appropriate to propose a re-test period or shelf life when stored at room temperature of up to twice but not more than 12 months beyond the period covered by long-term data, when the proposal is supported by the result of the analysis and relevant supporting data. For refrigerated chemical entities, where statistical analysis is performed, the proposed re-test period or shelf life can be up to one-and-a-half times, but should not be more than 6 months beyond, the period covered by long-term data.
如果长期数据可用于统计分析,但未进行分析,其外推程度应与数据不可用于统计分析时相同。然而,如果进行了统计分析,并且分析结果和相关支持性数据支持该建议,对于室温下贮藏的产品,建议的复检期或有效期可长达2倍,但不应超过长期数据涵盖时间后12个月。
对于进行统计分析的冷藏化学实体,建议的复检期或有效期可长达1.5倍,但不应超过长期数据涵盖时间后6个月。
13.2.7 Significant Change at Accelerated Condition
13.2.7 加速条件下的显著变化
Where significant change occurs at the accelerated condition, the re-test period or shelf life would depend on the storage condition (room temperature or refrigerated) and if stability data at an intermediate condition are available.
如果在加速条件下发生显著变化,复检期或有效期将取决于贮藏条件(室温或冷藏)以及中间条件下的稳定性数据是否可用。
13.2.7.1 Significant Change at Accelerated Condition (refrigerated storage) (Scenario C)
加速条件下的显著变化(冷藏)(场景C)
For refrigerated storage, if significant change occurs at the accelerated storage condition, the proposed re-test period or shelf life should be based on the long-term data and extrapolation is generally not considered appropriate. Intermediate conditions are also not considered applicable for products stored at refrigerated storage conditions. In addition, a re-test period or shelf life shorter than the period covered by long-term data could be proposed in a science- and risk-based manner. If the long-term data show variability, verification of the proposed re-test period or shelf life by statistical analysis can be appropriate.
对于冷藏条件,如果在加速条件下发生显著变化,建议的复检期或有效期应基于长期数据,一般不宜外推。中间条件也不适用于在冷藏条件下贮藏的产品。此外,可以以科学和基于风险的方式提出短于长期数据涵盖的时间周期的复检期或有效期。如果长期数据显示出变异性,则可通过统计分析对建议的复检期或有效期进行验证。
13.2.7.2 Significant Change at Accelerated Condition and Significant Change at Intermediate
Condition (room temperature storage) (Scenario D)
加速条件和中间条件(室温贮藏)下均有显著变化(场景D)
Where significant change occurs at both accelerated and the intermediate condition, the proposed re- test period or shelf life should be based on the long-term data and extrapolation is generally not considered appropriate. In addition, a re-test period or shelf life shorter than the period covered by long-term data could be proposed in a science- and risk-based manner. If the long-term data show variability, verification of the proposed re-test period or shelf life by statistical analysis can be appropriate.
如果在加速试验和中间条件均发生显著变化,建议的复检期或有效期应基于长期数据,通常不宜采用外推法。此外,可以以科学和基于风险的方式提出短于长期数据涵盖的时间周期的复检期或有效期。如果长期数据显示出变异性,则可通过统计分析对建议的复检期或有效期进行验证。
13.2.7.3 Significant Change at Accelerated Condition and No Significant Change at
Intermediate Condition (room temperature storage) (Scenario E)
加速条件下有显著变化,中间条件(室温贮藏)下无显著变化(场景E)
If there is significant change at accelerated condition but no significant change at the intermediate condition, extrapolation beyond the period covered by long-term data can be proposed; however, the extent of extrapolation would depend on whether long-term data for the attribute are amenable to statistical analysis.
如果在加速条件下有显著变化,但在中间条件下无显著变化,可以建议外推至长期数据涵盖时间周期之外;但外推的范围将取决于该属性的长期数据是否适合进行统计分析。
13.2.7.3.1 Data not amenable to statistical analysis (Scenario E.I)
数据不可用于统计分析(场景E.I)
When the long-term data for an attribute are not amenable to statistical analysis, the proposed re-test period or shelf life can be up to 3 months beyond the period covered by long-term data, if supported by relevant supporting data.
当某一指标的长期数据不适合进行统计分析时,如果有相关支持性数据支持,建议的复检期或有效期最长为长期数据涵盖时间后3个月。
13.2.7.3.2 Data amenable to statistical analysis (Scenario E.II)
数据可用于统计分析(场景E.II)
When the long-term data for an attribute are amenable to statistical analysis but no analysis is performed, the extent of extrapolation should be the same as when data are not amenable to statistical analysis.However, if a statistical analysis is performed, the proposed re-test period or shelf life can be up to one- and-half times, but should not be more than 6 months beyond, the period covered by long-term data, when backed by statistical analysis and relevant supporting data.
当一个指标的长期数据可用于统计分析,但未进行分析时,其外推程度应与数据不可用于统计分析时相同。但如果进行统计分析,在有统计分析和相关支持性数据支撑的情况下,建议的复检期或有效期可达1.5倍,但不应超过长期数据涵盖时间后6个月。
13.2.8 Extrapolation for Chemical Entities when Stored Frozen
13.2.8 冷冻保存时化学实体的外推
When a drug substance or product is stored frozen, with no observable or no statistically significant change over time for the available data of all quality attributes monitored at the recommended storage conditions or a minor change that remains well within the acceptance criteria, extrapolation may be considered based on appropriate prior knowledge and enhanced stability modelling (Annex 2 –Stability Modelling).
当原料药或制剂以冷冻方式保存时,在推荐的贮藏条件下监测的所有质量属性的可用数据随时间未观察到的变化或无统计学意义的变化,或仅出现微小的变化且仍在可接受标准范围内时,可以基于适当的先验知识和增强的稳定性建模进行外推(附录2-稳定性建模)。
13.2.9 Extrapolation for Biologicals
13.2.9 生物制品外推
Extrapolation beyond the period covered by available long-term primary stability data may be considered for a well characterised biological drug substance stored frozen, for which the quality attributes are known, and their corresponding criticality and residual risks evaluated to ensure patient safety. Extrapolation of drug substance shelf life should be limited to one and a half times the available long-term data from the primary stability batches to a maximum of 12 months beyond available long- term data, when justified. Justification should include a risk-based approach to fully support the proposed extrapolation, including data available on batches that have long term data to the end of the proposed shelf life that are analytically comparable to primary batches. Justification should also include statistical analysis (such as using linear regression with 95% confidence limit) of available long-term data on representative batches and primary stability batches to show no statistically significant or meaningful change over time. Any observable trend should also be justified. In addition, the risk assessment should take into consideration other aspects such as, knowledge of the molecule and its degradation profile, impact of degradation of the molecule on drug product, knowledge of the impact on stability due to the rate of freezing and thawing of the drug substance, the container/closure system,drug substance concentration and formulation to support the extrapolation.
对于冷冻保存且质量属性已知的表征良好的生物制品原液,可考虑在现有长期注册稳定性数据涵盖时间之外进行外推,并评估其相应的关键性和残余风险,以确保患者安全。在合理情况下,原液有效期的外推应限制在注册稳定性批次可用长期数据的1.5倍至可用长期数据后的最多12个月。证明应包括基于风险的方法,以充分支持建议的外推,包括具有至建议有效期结束的长期数据的批次的可用数据,这些数据在分析上与注册稳定性批次具有可比性。理由还应包括对代表性批次和注册稳定性批次的可用长期数据的统计分析(如使用95%置信限的线性回归),以显示随时间推移没有或有统计学意义的变化。任何可观察到的趋势也应合理。此外,风险评估应考虑其他方面,例如了解分子及其降解产物谱、分子降解对制剂的影响、了解原液的冻融速率对稳定性的影响、容器/密封系统、原液药浓度和剂型,以支持外推。
Alternative approaches can be proposed and justified for extrapolation and/or shelf life prediction based on appropriate prior knowledge and enhanced stability modelling (Annex 2 –Stability Modelling).
可以根据适当的先验知识和增强的稳定性建模(附录2-稳定性建模)提出外推和/或有效期预测的替代方案,并证明其合理性。
The general principles outlined here for drug substance extrapolation may be applicable to drug product extrapolation, however, due to increased risk, applicants are encouraged to seek agreement with regulatory authorities on the extrapolation proposal and accompanying justification that includes potential impact to patient safety and efficacy. Additionally, for biological drug products, applicants are encouraged to consider enhanced modelling techniques as described in Annex 2 – Stability Modelling.
此处概述的原液外推的一般原则可能适用于制剂外推,但由于风险增加,鼓励申请人与监管机构就外推方案和随附的理由(包括对患者安全和有效性的潜在影响)达成一致。此外,对于生物制剂,鼓励申请人考虑附录2-稳定性建模中所述的增强建模技术。
For biologicals and synthetics, when the proposed shelf life is extrapolated beyond available long-term data from primary stability studies, the primary stability studies should be continued post-approval to confirm the shelf life with long-term data. The ongoing monitoring/trending of stability data should be managed by the manufacturer’s PQS. The PQS should be capable of detecting and managing any confirmed changes in stability trend and out of specification results with appropriate corrective action and preventive actions (CAPA) as described in ICH Q10, relevant to any extrapolation being applied.
对于生物制品和化学合成实体,当建议的有效期外推至注册稳定性研究中可用的长期数据之外时,应在批准后继续进行注册稳定性研究,以使用长期数据确认有效期。稳定性数据的持续监测/趋势分析应由生产商的PQS进行管理。如在ICH Q10中描述的任何外推被引用,PQS应有能检测和管理在稳定性趋势中已经确认的变化和伴随适当纠正措施和预防措施(CAPA)的超标结果的能力。
13.3 Data Evaluation for Multi-factor, Full-design Studies
13.3 多因素、完整设计研究的数据评估
The stability of the drug product, or drug substance if applicable, could differ to a certain degree among different factor combinations in a multi-factor, full-design study, for example, products with different fill volumes or content and different container dimensions. Two approaches can be considered when analysing such data.
在多因素、完整设计研究中,制剂或原料药(如适用)的稳定性在不同因素组合之间可能存在一定程度的差异,比如在那些具有不同装量/含量以及不同容器尺寸的产品之间。在分析这些数据时,可以考虑两种方法。
To determine whether the data from all factor combinations (e.g., fill volume and container
dimensions such as vial size), support the proposed shelf life for each combination of drug
product presentation.
确定所有因素组合(比如装量和小瓶尺寸等容器尺寸)的数据是否支持每种制剂组合的建议有效期。
To determine whether the data from different factor combinations can be combined for an
overall estimate of a single shelf life that applies to each presentation.
确定不同因素组合的数据是否可以合并,以总体估计适用于每种呈现方式的单一有效期。
A statistical model that includes all appropriate factors and factor combinations may be constructed and the shelf life should be estimated for each factor and for all factor combinations to support the product shelf life.
可构建包括所有适当因素和因素组合的统计模型,并估算每个因素和所有因素组合的有效期,以支持产品有效期。
If all shelf lives estimated by the aforementioned statistical model are longer than the proposed shelf life, further model building is considered unnecessary, and the proposed shelf life will generally be appropriate for all combinations of factors. The stability data from different factors should not be combined unless supported by scientific understanding and statistical testing.
如果上述统计模型估计的所有有效期均长于建议的有效期,则认为无需进一步建立模型,建议的有效期通常适用于所有因素组合。除非有科学理解和统计检验的支持,否则来自不同因素的稳定性数据不应合并。
13.3.1 Testing to Combine Batch Data per Individual Combination
13.3.1 用于每个单独组合批次数据的合并测试
If each factor combination is considered separately, the stability data can be statistically tested to combine those batch data for each individual combination. The shelf life for each non-batch factor combination can be estimated separately by applying the procedure described for single factor, full design (Refer to Annex 2, Section A2-1 – Statistical Evaluation of Stability Data from Single or Multi-Factor Study Designs). For example, for a drug product available in two strengths and four container sizes, eight sets of data from the 2 x 4 strength-size combinations can be analysed and eight separate shelf lives should be estimated accordingly. For a single shelf life across the strengths and container sizes, the shortest (worst-case) estimated shelf life among all factor combinations should become the
shelf life for the product. However, this approach does not consider all the available data from all factor combinations, thus generally resulting in shorter shelf lives than the approach that combines batches for all factors and factor combinations.
如果单独考虑每个因素组合,则可以对稳定性数据进行统计检验,以合并每个单独组合的批次数据。每种非批次因素组合的有效期可通过应用单因素、完整设计描述的方法单独估计(见附录2第A2-1节“单一或多因素研究设计稳定性数据的统计评估”)。例如,对于一种有2种规格和4种容器尺寸的制剂,可以分析2×4规格尺寸组合的8组数据,并相应地估计8个单独的有效期。对于适用于不同规格和容器大小的单一有效期,所有因素组合中最短(最坏情况)的预计有效期应与产品有效期一致。然而,这种方法没有考虑所有因素组合的所有可用数据,因此通常会导致相较于采用合并所有因素及其组合批次的方法,这种方法的有效期更短。
13.3.2 Testing to Combine Data for All Factors and Factor Combinations
13.3.2 综合所有因素和因素组合数据的测试
If the stability data are tested to combine all factors and factor combinations and the results show that the data can be combined, a single shelf life across all combinations and longer than that estimated based on individual factor combinations may be proposed. The shelf life is longer because the width of the confidence limit(s) for the mean will become narrower as the amount of data increases when batches, strengths, container sizes and/or fills, etc. are combined into a single analysis of covariance (e.g., ANCOVA).
如果稳定性数据测试结果表明,能够将所有因素和因素组合的数据进行结合,则可以提议在所有组合中采用统一的有效期,并且该有效期可能长于根据单个因素组合估算的有效期。有效期更长的原因是,当批次、规格、容器尺寸和/或装量等被合并为单一的协方差分析(比如ANCOVA)时,随着数据量的增加,均值的置信区间会变得更窄。
Analysis of covariance (e.g., ANCOVA) can be employed to test the difference in slopes and intercepts of the regression lines among factors and factor combinations. The purpose of the procedure is to determine whether data from multiple factor combinations can be combined for the estimation of a single shelf life that could apply to all 8 presentations for the previous example (refer to Section 13.3.1- Testing to Combine Batch Data per Individual Combination).
协方差分析(比如ANCOVA)可用于检验因素和因素组合之间回归线的斜率和截距的差异。该程序的目的是确定来自多个因素组合的数据是否可以合并,用于估算一个适用于前一个示例中所有8种呈现方式的统一有效期(见第13.3.1节“用于每个单独组合批次数据的合并测试”)。
The full statistical model should include the y-intercept and slope terms for all main effects and interaction effects and a term reflecting the random error of measurement. If it can be justified that the higher order interactions are very small, there is generally no need to include these terms in the model.In cases where the analytical results at the initial time point are obtained from the dosage form prior to its packaging, the effect of container is taken into account in each measure as comparisons are made to the initial time point analysed prior to packaging.
完整统计模型应包括所有主要作用和交互作用的y轴截距和斜率项,以及反映测量随机误差的项。如果可以证明高阶相互作用非常小,则通常无需在模型中包含这些项。如果初次时间点的分析结果是从包装前的剂型中获得的,则在每次测量时都要考虑容器的影响,与包装前分析的初次时间点进行比较。
The tests to combine data should be specified to determine whether there are statistically significant differences among factors and factor combinations. Generally, the statistical tests for covariance should be performed in a proper order such that the slope terms are tested before the intercept terms and the interaction effects are tested before the main effects. For example, the tests can start with the slope and then the intercept terms of the highest order interaction and proceed to the slope and then the intercept terms of the simple main effects. The most reduced model, obtained when all remaining terms are foun to be statistically significant, can be used to estimate the shelf life.
应规定合并数据的测试,以确定各因素和因素组合之间是否存在统计学上的显著差异。一般而言,协方差的统计检验应以适当的顺序进行,从而使斜率项在截距项之前检验,交互作用在主效应之前检验。比如说,检验可以从最高阶相互作用的斜率和截距项开始,然后到简单主效应的斜率和截距项。当所有剩余项均具有统计学意义时,可使用最简化模型来估计有效期。
All tests should be conducted using appropriate levels of significance (refer to Annex 2 – Stability Modelling). Typically, a significance level of 0.25 can be used for batch-related terms, and a significance level of 0.05 can be used for non-batch-related terms. If the tests show that the data from different factor combinations can be combined, the shelf life can be estimated according to the procedure described for a single batch (refer to Section 13.2.1 – Linear Regression for an Individual Batch), using the combined data.
所有检验均应以适当的显著性水平进行(见附录2-稳定性建模)。通常情况下,0.25的显著性水平可用于批次相关术语,0.05的显著性水平可用于非批次相关术语。如果检验结果表明不同因素组合的数据可以合并,则可根据单个批次描述的程序(见第13.2.1节“单一批次的线性回归”),使用合并后的数据估计有效期。
If the tests show that the data from certain factors or factor combinations should not be combined, then a single shelf life can be estimated based on the shortest estimated shelf life among all levels of factors and factor combinations remaining in the model.
After model selection and implementation, model lifecycle consideration should be considered per Annex 2 - Stability Modelling, Section 2.7 – Risk Management and Model Lifecycle Considerations.
如果检验表明某些因素或因素组合的数据不应合并,则可以根据模型中剩余的所有水平的因素和因素组合中最短的估计有效期来估算单一的有效期。
模型选择和实施后,应根据附录2-稳定性建模第2.7节“风险管理和模型生命周期考虑因素”考虑模型生命周期因素。
13.4 Data Presentation
13.4 数据呈现
The applicant should follow ICH M4Q for data presentation expectations. In general, for stability data,data for all attributes should be presented in an appropriate format (e.g., tabular, graphical, narrative) and an evaluation of such data. The values of quantitative attributes at all time points should be reported as measured and as calculated to support the label claim, where applicable. If a statistical analysis is performed, the procedure used and the assumptions underlying the model should be stated and justified.
申请人应遵循ICH M4Q的数据呈现预期。一般而言,对于稳定性数据,应以适当格式(如表格、图形、叙述性)对所有属性数据进行呈现,并对这些数据进行评估。在适用的情况下,所有时间点的定量属性值应报告为测量值和计算值,以支持标签声明。如果进行了统计分析,应说明并证明所使用的程序和模型的假设。
14 LABELLING
14 说明书及标签
Guidance for labelling and storage statements for drug substances and drug products are provided below.
原料药和制剂的说明书及标签的贮藏说明指导如下。请注意,在适用的情况下,相同的原则也应适用于中间产品的贮藏。
Note that the same principles should be applied to stored intermediates when applicable.
A storage statement should be established for the labelling based on the evaluation of stability data with respect to the climatic zone where the drug substance and/or drug product are intended to be stored, shipped, or used. When applicable, storage statements should reflect information related to the in-use period and storage conditions. It is recommended that an appropriate temperature range be included on the label. Terms such as “ambient conditions’ or “room temperature” should be avoided on the label.
应根据原料药和/或制剂拟贮藏、运输或使用的气候带的稳定性数据评估,为标签制定贮藏说明。在适用的情况下,贮藏说明应反映与使用中允许时限和暂存条件相关的信息。建议在说明书及标签上注明适当的温度范围。说明书及标签上应避免使用“环境条件”或“室温”等术语。
Where applicable, specific instructions should be provided within the labelling, particularly for drug substances, intermediates and drug products that cannot tolerate freezing and thawing, exposure to light or humidity. Additional information may be included on the label for drug products with an established short-term storage condition (refer to Section 10 – Short-Term Storage Conditions).
在适用的情况下,应在说明书及标签中提供具体说明,特别是对于不能耐受冻融、暴露于光照或湿度的原料药、中间产品和制剂。对于具有既定短期条件的制剂,说明书及标签上可能包含其他信息(见第10节“短期条件”)。
There should be a direct link between the label storage statements and the demonstrated stability. An expiration date/re-test date, derived from the stability information, should be displayed on the container closure system labelling, as appropriate.
说明书及标签贮藏说明和已证明的稳定性之间应该有直接联系。根据稳定性信息制定的失效日期/复验日期应酌情显示在容器密封系统的标签上。
14.1 Excursins Outside of a Labelling Claim
14.1 超出说明书及标签要求的偏离
The quality attributes of pharmaceutical drug substances and drug products can be impacted by the extent of the environmental factors experienced during handling, transport, and storage. Those impacts should be evaluated and specified instructions may be provided on the product labelling.
原料药和制剂的质量属性可能会受到搬运、运输和贮藏过程中环境因素的影响。应评估这些影响,并在药品说明书及标签上提供具体说明。
Transient temperature excursions outside of the label storage conditions, may be acceptable if justified and supported by stability data. An assessment of the risk and impact of handling, transport, and storage excursions outside the label claim at various stages throughout the overall supply chain requires a comprehensive knowledge of the supply chain and an understanding of a drug substance and drug product’s stability profile. Data from stability studies, including accelerated studies, stress testing (Refer to Section 2 – Development Studied Under Stress and Forced Conditions), or transport simulation studies (when appropriate) can be used to evaluate the effects of an excursion on the drug substance or drug product. Additionally, statistical evaluation or modelling can be leveraged to evaluate the impact of a storage condition excursion, provided sufficient knowledge of the degradation pathway is available and fits an appropriate model. Each excursion should be documented and handled within the corresponding quality management system or appropriate risk assessment.
如果合理且有稳定性数据支持,则标签贮藏条件之外的瞬时温度偏离或许是可以接受的。在整个供应链的各个阶段,对超出标签声明的搬运、运输和贮藏偏离的风险和影响的评估需要对供应链有充分理解,并了解原料药和制剂的稳定性特征。稳定性研究,包括加速稳定性研究、影响因素试验(见第2节“影响因素和强制降解条件下的开发研究”)或模拟运输研究(如适用)的数据可用于评估偏离对原料药或制剂的影响。此外,如果对降解途径有足够的了解并符合适当的模型,则可以利用统计评价或建模来评价贮藏条件偏离的影响。应在相应的质量管理体系或适当的风险评估中记录和处理每次偏离情况。
15 STABILITY CONSIDERATIONS FOR COMMITMENTS AND PRODUCTLIFECYCLE MANAGEMENT
15 关于承诺和产品生命周期管理的稳定性考虑因素
Consistent with ICH Q8, the product lifecycle includes all phases in the life of a drug substance and drug product from the initial development through the marketing until the product’s discontinuation.
与ICH Q8一致,产品生命周期包括原料药和制剂从初始开发到上市直至产品终止的所有阶段。
Lifecycle management in the context of stability includes initial stability testing and re-test period and shelf life determination, ongoing (annual) stability testing, and stability studies supporting post-approval changes or commitments over a product’s lifecycle. This also includes the introduction of new dosage forms or new strengths/concentrations. Commitment stability studies include studies to confirm the initially proposed re-test period/shelf life for commercial manufacture. This section also provides guidance on stability studies necessary to support the product lifecycle after an initial re-test period or shelf life has been established in the regulatory submission. While guidance in this section is focused on product lifecycle management of drug substances and drug products, general principles may also apply to intermediates that require studies to support re-test period/shelf life or holding times.
In cases where data from commitment stability studies fall outside the acceptance criteria, as confirmed through quality investigation, the stability commitment should include a proposed action to the competent authority in accordance with regional requirements.
稳定性背景下的生命周期管理包括初始稳定性试验、复验期和有效期确定、持续(年度)稳定性试验、和支持产品生命周期内批准后变更或承诺的稳定性研究。其中还包括引入新的剂型或新规格/浓度。稳定性承诺研究包括对最初建议的用于商业化生产的复验期/有效期的确认研究。本节还提供了在监管申报中确定初始复验期或有效期后,支持产品生命周期所需的稳定性研究指导。虽然本节的指导侧重于原料药和制剂的产品生命周期管理,但一般原则也可适用于需要研究以支持复验期/有效期或保持时限的中间产品。
如果承诺稳定性研究的数据超出可接受标准,经质量调查确认后,则稳定性承诺应包括根据区域要求向监管部门提出的建议措施。
15.1 Commitment Stability Studies
15.1 稳定性研究承诺
Commitment stability studies are conducted under the accelerated, intermediate, or long-term storage conditions (as applicable) to establish or confirm the initial re-test period or shelf life. Where the primary stability studies for a drug substance or drug product do not cover the proposed re-test period or shelf life period granted at the time of initial approval, a commitment should be made to continue the stability studies to confirm the proposed re-test period or shelf life. If applicable, data supporting the claim that manufacturing scale does not impact stability of the product should be provided for regulatory assessment. When all the batches used in the primary stability studies are production batches and stability data cover proposed re-test period and/or shelf life, a post-approval commitment is considered unnecessary. Otherwise, one of the following commitments should be made:
在加速、中间或长期条件下(如适用)进行稳定性研究承诺,以建立或确认初始复验期或有效期。原料药或制剂的注册稳定性研究未涵盖首次批准时所建议的复验期或有效期时,应承诺继续进行稳定性研究以确认所建议的复验期或有效期。在适用的情况下,应提供支持生产规模不影响产品稳定性这一说法的数据,以供监管评估。当注册稳定性研究使用的所有批次均为生产批次,且稳定性数据涵盖建议的复验期和/或有效期时,则认为无需进行批准后承诺。否则,应作出下列承诺之一:
If the regulatory submission includes long-term data from stability studies less than the re-test period/shelf life for at least three production batches, a commitment should be made to continue these studies through the proposed re-test period/shelf life.
如果注册申报的稳定性研究长期数据少于三个生产批次的复验期/有效期,则应承诺在建议的复验期/有效期内继续进行这些研究。
If the regulatory submission includes data from stability studies on fewer than three production batches, a commitment stability study should be conducted to generate stability data on at least three production scale batches in total. Commitment stability studies under the long-term storage conditions should be initiated or continued though the proposed re-test period and/or/ shelf life and, if applicable, under the accelerated storage conditions through to 6 months.
如果注册申报包含少于三个生产批次的稳定性研究数据,则应进行承诺稳定性研究,以生成总共至少三个生产规模批次的稳定性数据。应开始或继续进行长期条件下的承诺稳定性研究,直至达到建议的复验期和/或有效期;在适用的情况下,还应进行加速条件下6个月试验。
For synthetics, if the regulatory submission does not include stability data on production
batches, a commitment stability study should be conducted to generate stability data on at least three production scale batches. Commitment stability studies under the long-term storage conditions should be initiated and continued through the proposed re-test period and/or shelf life and, if applicable, under the accelerated storage conditions through to 6 months.
对于化学合成药物,如果注册申报期间不包括生产批次的稳定性数据,则应进行稳定性承诺研究,并补充至少三个生产规模批次的稳定性数据。应开始并继续进行长期条件下的稳定性承诺研究,直至达到建议的复验期和/或有效期;在适用的情况下,还应进行加速条件下6个月试验。
The commitment stability study protocol should be the same as that for the primary stability study, unless otherwise scientifically justified. Continuation or application of new bracketing or matrixing approaches in the commitment stability studies for the stability commitment should also be justified as discussed in Annex 1 - Reduced Stability Protocol Design.
除非另有科学依据,否则稳定性承诺研究方案应与注册稳定性研究相同。对于在稳定性承诺研究中继续或采用新的括号法或矩阵设计,应按照附录1-简化稳定性方案设计中的讨论进行充分论证。
15.2 Ongoing Stability Studies
15.2 持续稳定性研究
Ongoing stability studies are conducted under long-term storage conditions on an annual basis to ensure the consistency of stability related quality attributes at the commercial storage conditions over the product lifecycle. These studies also allow for the monitoring of the stability characteristics and examine trends in the stability data to confirm the appropriate storage conditions relevant for the product and to confirm a re-test period or a shelf life.
每年在长期条件下进行持续稳定性研究,以确保在商业化贮藏条件下,产品生命周期内稳定性相关质量属性的一致性。这些研究还允许监测稳定性特征并检查稳定性数据的趋势,以确认与产品相关的适当贮藏条件,并确认复验期或有效期。
In accordance with the general principles in ICH Q7, at least one production batch of the drug substance and one production batch of each strength of the drug product covering the container closure systems should be added to the ongoing stability program per year (unless none is produced that year). Ongoing stability studies are generally managed within the PQS unless a regulatory authority expects additional submission of the information and data. Each production site should maintain an ongoing stability programme in accordance with GMPs. Reduced designs (as discussed below and in Annex 1- Reduced Stability Protocol Design) can be applied where justified.
根据ICH Q7中的一般原则,每年至少应将一个原料药生产批次和覆盖容器密封系统的每种规格药品一个生产批次增加到持续稳定性计划中(除非当年未生产)。持续稳定性研究通常在PQS内进行管理,除非监管机构要求额外提交信息和数据。每个生产场地应根据GMP保持持续稳定性计划。在合理的情况下,可采用简化设计(如下文和附录1-简化稳定性方案设计中所述)。
Ongoing stability studies are not required to align with the primary stability protocol; however, testing should continue through to the end of the re-test period or shelf life. As product knowledge is gained,the applicant may consider removal of testing of attributes not related to stability and/or reduce testing timepoints based on risk assessment as detailed in Section 3 - Stability Protocol Design. Reductions,including bracketing and/or matrixing approaches, based on stability knowledge and risk assessment should be justified in the regulatory submission, where applicable, as detailed in Annex 1 (Reduced Stability Protocol Design). Reduced protocol designs applied in the original regulatory submission should be followed until there is a change in configuration (e.g., strength/concentration). Any change
in the reduced design post-approval should be evaluated for its impact to the product quality prior to modifying the annual stability protocol. While the testing intervals listed during product development may be appropriate in the pre-approval stage, reduced testing may be appropriate after approval where data are available that demonstrate adequate and consistent stability. Where data exist that indicate the stability of a product is not compromised, the applicant is encouraged to propose and justify, where applicable, a protocol which supports the reduction or elimination of specific testing (e.g., 9-month testing interval) or certain attributes (e.g., orthogonal testing) for post-approval, long-term studies.
持续稳定性研究不需要与注册稳定性方案保持一致,但试验应持续到复验期或有效期结束。随着产品知识的获得,申请人可以考虑删除与稳定性无关的属性测试和/或根据第3节“稳定性方案设计”中详述的风险评估减少测试时间点。基于稳定性知识和风险评估的简化措施(包括括号法和/或矩阵设计),应在注册提交中说明其合理性,详见附录1-简化稳定性方案设计。应遵循原始注册提交文件中应用的简化方案设计,直到配置发生变化(比如规格/浓度变化)。在修改年度稳定性方案之前,应评估批准后简化设计的任何变更对产品质量的影响。虽然产品开发过程中列出的试验间隔在批准前阶段可能是合适的,但如果有数据证明稳定性充分且一致,则在批准后可适当简化检测。如果有数据表明产品的稳定性没有受到影响,鼓励申请人提出并证明一个方案(如适用),支持批准后长期研究中简化或取消专属性检测(如9个月的试验间隔)或某些属性(如正交测试)。
15.3 Product Lifecycle Stability Studies
15.3 产品生命周期稳定性研究
Product lifecycle stability studies are conducted under the accelerated, intermediate, or long-term storage conditions (as applicable) to support product lifecycle changes by assessing whether the change has an impact on any stability related quality attributes of the commercial drug substance or product under the labelled storage, handling and use conditions. A risk assessment should be conducted (refer to Section 3 – Stability Protocol Design and Annex 1 – Reduced Stability Protocol Design) and can be used to justify the change and determine the need and extent of studies required to support changes after approval in compliance with regional requirements. A post-approval change could fall into one of the following scenarios that are based on the nature and impact of the change, stability data requirements and where the re-test/shelf life establishment could change:
在加速、中期或长期条件下(如适用)进行产品生命周期稳定性研究,通过评估变更是否对市售原料药或产品在标示的贮藏条件、搬运和使用条件下的任何稳定性相关质量属性产生影响,以支持产品生命周期变更。应进行风险评估(见第3节“稳定性方案设计”和附录1-简化稳定性方案设计),可用于证明变更的合理性,并确定在按照区域要求批准后支持变更所需的研究的需求和范围。根据变更的性质和影响、稳定性数据要求以及复验期/有效期确定可能发生变化的情况,批准后变更可分为以下几种情况之一:
Scenario 1: A stability risk assessment indicates the proposed changes will not have an impact on the stability profile (e.g., change to a comparable analytical procedure, change in outside cap colour).Stability data in this case is unnecessary and the re-test period or shelf life will not be re-established.Maintained product stability would be confirmed as part of the Ongoing Stability Programme.
场景1:稳定性风险评估表明建议的变更不会对稳定性特征产生影响(比如类似分析方法的变更、外盖颜色的变更)。在这种情况下,不需要稳定性数据,也不会重新建立复验期或有效期。维持产品稳定性将作为持续稳定性计划的一部分进行确认。
Scenario 2: The proposed changes may potentially impact the stability profile (e.g., manufacturing process change, change in formulation). A stability study, a stability risk assessment, or a combination thereof may be appropriate to support this change. The risk assessment process may include a well-designed study to determine whether additional formal stability studies or other supportive stability studies are necessary. The assessment should establish whether the re-test period/shelf life and storage condition may be maintained or if they should be re-established.
场景2:建议的变更可能会影响稳定性特征(如生产工艺变更、剂型变更)。稳定性研究、稳定性风险评估或其组合可能适用于支持该变更。风险评估过程可包括精心设计的研究,以确定是否需要额外的正式稳定性研究或其他支持性稳定性研究。评估应确定是否可以维持复验期/有效期和贮藏条件,或者是否应该重新设定。
- If the proposed changes have a demonstrated impact that can reduce or extend the re-test period/shelf life based on the preliminary stability results, then a re-test period/shelf life
and storage condition may need to be re-established per recommendations in Section 3 -
Stability Protocol Design through Section 7 - Storage Conditions.
- 根据初步稳定性结果,如果建议的变更具有能够缩短或延长复验期/有效期的影响,则可能需要根据第3节“稳定性方案设计”至第7节“贮藏条件”中的建议重新确定复验期/有效期和贮藏条件。
- If the proposed change is expected to have a low impact but formal stability studies are
warranted based on preliminary data and risk assessment, a commitment should be made
to continue these stability studies through the re-test period/shelf life and the re-test period
or shelf life does not need to be re-established.
- 如果建议的变更预期影响较小,但根据初步数据和风险评估,需要进行正式稳定性研究,则应承诺在复验期/有效期内继续进行这些稳定性研究,并且不需要重新建立复验期或有效期。
- If the proposed change is demonstrated through the risk assessment and/or a well-designed stability study (including analytical comparability according to ICH Q5E for biological products), to not impact the re-test period or shelf life, then this assessment and/or data may be used to justify that formal stability studies are not needed to retain the current re-test period or shelf life (e.g., change in compendial excipient supplier).
- 如果建议的变更通过风险评估和/或精心设计的稳定性研究(包括依据ICH Q5E的生物制品分析可比性)表明不会影响复验期或有效期,则该评估和/或数据可用于证明不需要进行正式稳定性研究来保留当前的复验期或有效期(比如药典级辅料供应商的变更)。
- If a risk assessment or an initial set of stability results do not allow for an understanding of the impact to the re-test period/shelf life, the re-test period/shelf life and storage condition
may need to be re-established based on the post-change stability data.
- 如果无法通过风险评估或最初的一组稳定性结果理解对复验期/有效期的影响,则可能需要根据变更后的稳定性数据重新建立复验期/有效期和贮藏条件。
- Product lifecycle stability studies intended to extend the re-test period or shelf life should align with the principles outlined for primary stability (e.g., for setting re-test period/shelf life). Justification should be provided when a shelf life reduction is proposed as a post-approval change. This justification should only be based on scientific reasons.
- 旨在延长复验期或有效期的产品生命周期稳定性研究应符合注册稳定性的原则(比如设定复验期/有效期的原则)。当建议将有效期缩短作为批准后变更时,应提供相关依据。且这一依据只能基于科学理由。
In most circumstances, stability evaluation is generally expected in the context of the specific change and should include assessment of impact on drug substance, intermediate and/or final drug product.Additional scientific, risk-based considerations and approaches for identifying stability-related quality attributes, use of appropriate tools to evaluate the impact of the intended change and developing strategies for confirmatory stability studies supporting stability for post-change material are included in ICH Q12, Chapter 9 (Stability Data Approaches to Support the Evaluation of CMC Changes) and recommendations for post-approval changes. For biologicals, after successful demonstration of analytical comparability according to ICH Q5E including the stability profile, the shelf life of the pre-
change material can be assigned to the post-change material. If successful demonstration of analytical comparability is not achieved, additional stability studies would be needed.
在大多数情况下,稳定性评估通常在特定变更的背景下进行,并应包括对原料药、中间产品和/或成品制剂影响的评估。ICH Q12第9章(中间产品加工和贮藏时间的稳定性考虑因素)和批准后变更建议中包含了其他科学的、基于风险的考虑因素和方法,以确定稳定性相关的质量属性、使用适当的工具来评估预期变更的影响,并制定支持变更后物料稳定性的确证稳定性研究策略。对于生物制品,在根据ICH Q5E成功证明分析可比性(包括稳定性特征)后,可将变更前材料的有效期分配给变更后材料。如果未能成功证明分析可比性,则需要进行额外的稳定性研究。
In some instances, a stability protocol may include additional time points beyond a proposed shelf life to allow shelf life extensions in the future (e.g., to avert supply management issues). An extension of the approved shelf life based on acceptable stability data from a minimum of 3 production or primary batches may be submitted to allow a longer shelf life.
在某些情况下,稳定性方案可能包括超出建议有效期的额外时间点,以便将来可以延长有效期(比如避免供应管理问题)。根据至少3个生产或注册稳定性批次的可接受稳定性数据,可延长批准的有效期以获得更长的有效期。
The applicant should apply an appropriate stability strategy that demonstrates the established re-test period/shelf life and storage conditions are still accurate. In such cases, an appropriate stability strategy may include:
申请人应采用适当的稳定性策略,证明其建立的复验期/有效期和贮藏条件仍然准确。在这种情况下,适当的稳定性策略可包括:
A targeted stability study that focuses on the potentially impacted stability related quality
attributes and re-test period/shelf life limiting attributes.
一项有针对性的稳定性研究,重点关注可能受影响的稳定性相关质量属性和复验期/有效期限制性属性。
The use of comparative accelerated/stress and/or predictive stability studies (e.g., modelling,including extrapolation, or stability bridging study for biological product) to demonstrate the understanding from the process/product change.
使用比较加速/影响因素研究和/或预测稳定性研究(比如建模、外推,或生物制品的稳定性桥接研究)来证明对工艺/产品变更的充分理解。
A risk assessment demonstrating that an understanding of the impact to any stability related
quality attributes can support limited real-time data for post-change material while claiming
the same re-test period/shelf life as the pre-change material.
风险评估表明,对任何稳定性相关质量属性影响的充分理解可以支持变更后材料的有限实时数据,同时声称与变更前材料具有相同的复验期/有效期。
A full evaluation of stability related quality attributes through long-term studies. This may be
necessary when the impact of the change is not well understood or demonstrated.
通过长期研究对稳定性相关的质量属性进行全面评价。当变更的影响没有得到很好的理解或证明时,这一评价可能是必要的。
Reduced protocol designs may be applied for drug products with multiple commercial presentations where stability performance is generally well understood. For example, a worst-case approach may be applied to products with multiple bottle configurations, where the configuration with the highest moisture vapor transmission rate (MVTR) is selected for evaluation (refer to Annex 1 – Reduced Stability Protocol Design). Reduced protocol design considerations may also apply to photostability or in-use studies supporting changes such as primary/secondary packaging or in-use and should follow the same considerations as discussed above and in Section 8 - Photostability and Section 11 In-Use Stability.
简化方案设计可应用于具有多种商业表现的制剂,其中稳定性性能通常已得到充分理解。例如,在具有多种瓶装的产品中,可以采用最坏情况方法,在其中选择具有最高水蒸气透过率(MVTR)的包装进行评估(见附录1-简化稳定性方案设计)。简化方案设计考虑因素也可适用于支持变更(如内包装/次级包装或使用期间)的光稳定性或使用期间研究,并应遵循上述以及第8节“光稳定性”和第11节“使用中稳定性”中讨论的相同考虑因素。
If specific tests or timepoints from the primary stability studies had been removed for the ongoing stability protocol, these may need to be restored for the stability studies used to support a post-approval change.
如果持续稳定性方案删除了注册稳定性研究中的特定检测或时间点,则可能需要在支持批准后变更的稳定性研究中恢复这些检测项目或时间点。
15.4 Stability Studies to Support New Dosage Forms and New Strengths/Concentrations
15.4 支持新剂型和新规格/浓度的稳定性研究
This section addresses the recommendations on what should be submitted regarding stability of a new dosage form or a new strength/concentration by the owner of the original regulatory submission. A new dosage form or strength/concentration contains the same drug substance as included in the existing, approved drug product. Within scope of a new dosage form are new products with different administration route (e.g., oral to parenteral, intravenous to subcutaneous), new specific functionality/delivery systems (e.g., immediate release tablet to modified release tablet, lyophilised to liquid product) and different dosage forms of the same administration route (e.g., capsule to tablet, solution to suspension, vial to prefilled syringe).
本节讨论了关于新剂型或新规格/浓度的稳定性的建议,这些建议应由原注册申报资料的所有者提交。新剂型或规格/浓度含有与现有已获批制剂相同的原料药。在新剂型的范围内,具有不同给药途径(如从口服到胃肠外给药、从静脉注射到皮下注射)、新的特定功能/递送系统(如从常释片剂到缓释片剂、从冻干到液体产品)和相同给药途径的不同剂型(如胶囊到片剂、从溶液到混悬液、从药瓶到预填充式注射器)。
Stability protocols for new dosage forms or new strengths/concentrations should generally follow the guidance for primary stability studies (refer to Table 1). In certain justified cases, based on prior knowledge and an established stability profile, a science- and risk-based, reduced stability protocol at submission may be acceptable (e.g., 6 months accelerated and 6 months long term data for a new dosage form for a synthetic chemical entity per Table 1). In cases where the existing commercial data are relevant to the shelf life of the new dosage form or the new strength/concentration, a risk assessment with an appropriate justification and additional supporting information (e.g., predictive data, comparative bridging data and/or prior knowledge) should be provided. In these cases, a commitment stability study would also be expected in accordance with the principles discussed in Section 15.1 – Commitment Stability Studies.
新剂型或新规格/浓度的稳定性方案通常应遵循注册稳定性研究指导(见表1)。在某些合理的情况下,基于先验知识和既定的稳定性特征,在提交时基于科学和风险的简化稳定性方案是可以接受的(比如根据表1,化学药物实体新剂型的6个月加速数据和6个月长期数据)。
如果现有商业数据与新剂型或新规格/浓度的有效期相关,则应提供风险评估,并提供适当的依据和其他支持信息(比如预测性数据、比较性桥接数据和/或先验知识)。在这些情况下,根据第15.1节“承诺稳定性研究”中讨论的原则,预期也将进行承诺稳定性研究。
16 GLOSSARY
16 术语表
Accelerated Studies: Testing conducted on drug substance and drug product that have been stored under conditions intended to increase the rate of physical, chemical and/or biochemical change (temperature and when applicable humidity), over a defined time period. These data can be used to gain product knowledge and to support extrapolation, re-test period or shelf life determination and to evaluate the impact of excursions outside the label storage conditions.
加速稳定性研究:在规定时间内,对贮藏条件(旨在提高物理、化学和/或生化变化率(温度和湿度,如适用))下的原料药和制剂进行的试验。这些数据可用于获取产品知识,支持外推、复验期/有效期的确定,并评估偏离说明书/标签上的贮藏条件的影响。
AI-ML: Artificial Intelligence Machine Learning
AI-ML: 使用人工智能机器学习
ANCOVA: Analysis of covariance
ANCOVA: 协方差分析
ATMP: Advanced Therapy Medicinal Products Container Closure System: The sum of packaging components that together contain and protect the dosage form. This includes primary packaging components and secondary packaging components, if the latter are functional (e.g., combination of a drug product with a medical device) or intended to provide additional protection to the drug product. A packaging system is equivalent to a container closure system. For the drug substance the container closure system is the packaging proposed for storage and distribution.
ATMP: 先进治疗产品容器密封系统:用于盛装和保护制剂的包装组件的总和。包括内包装组件和次级包装组件,如果后者是功能性的(比如药械组合)或旨在为制剂提供额外保护。一个包装系统视为一个容器密封系统。对于原料药,容器密封系统是用于贮藏和分销的包装。
Commitment stability studies: Stability studies conducted under the accelerated, intermediate, or long-term storage conditions (as applicable) to establish or confirm the initial re-test period or shelf life in accordance with a commitment in the regulatory submission.
稳定性研究承诺:在加速、中间条件或长期条件下(如适用)进行的稳定性研究,用以根据注册申报中的承诺建立或确认初始复验期或有效期。
CAPA: Corrective and Preventive Actions (ICH Q12)
CAPA: 纠正措施和预防措施 (ICH Q12)
CM: Continuous Manufacturing (ICH Q13)
CM: 连续制造(ICH Q13)
CQA: Critical Quality Attributes (ICH Q8)
CQA: 关键质量属性(ICH Q8)
Degradation Product: Molecular variants or impurities resulting from chemical or biochemical changes in the desired product or product-related substances brought about over time and/or by the action of, e.g., light, temperature, pH, water, or by reaction with an excipient and/or the container closure system and/or device component. Such changes may occur as a result of manufacture and/or storage (e.g., hydrolysis, deamidation, oxidation, aggregation, proteolysis). Degradation products may be either product-related substances or product-related impurities.
降解产物:由于预期产品或与产品相关物质随时间推移及/或因光照、温度、pH值、水分或与辅料及/或容器密封系统及/或包装组件的反应作用下产生的化学或生化变化而生成的分子变体或杂质。这种变化可能是由于生产和/或贮藏(例如,水解、脱酰胺、氧化、聚集、蛋白水解)而发生。降解产物可能是产品相关物质或产品相关杂质。
DS: Drug Substance
DS: 原料药(生物制品也称原液)
DP: Drug Product
DP: 制剂
Full design stability protocol: A protocol which includes at least three batches of the drug substance or at least three batches of each strength or concentration of the drug product covering the container closure systems for every combination of all design factors and tested at all time points.
完整稳定性设计方案:在所有时间点纳入并检测至少三批原料药或每种规格/浓度的制剂(涵盖所有设计因素的每种组合的容器密封系统)的方案。
Formal Stability Studies: Primary, commitment, ongoing or product lifecycle stability studies
conducted under the accelerated, intermediate, or long-term storage conditions (as applicable) to establish or confirm a re-test period or a shelf life.
正式稳定性研究:在加速条件、中间条件或长期条件下(如适用)进行的所有注册、承诺、持续或产品生命周期稳定性研究,用于建立或确认复验期/有效期。
GMP: Good manufacturing practice
GMP:生产质量管理规范
IgG: Immunoglobulin G Impermeable Container: Containers that provide a permanent barrier to the passage of gases or solvents, e.g., sealed aluminium tubes for semi-solids, sealed glass ampoules for solutions and aluminium/aluminium blisters for solids.
IgG: 球蛋白G非渗透性容器:能对气体或溶剂通透设置永久性屏障的容器,如半固体密封铝管、溶液剂的密封玻璃安瓶和固体铝/铝泡罩。
Impurity: Any component of the drug substance or drug product which is not the synthetic chemical or biological entity defined as the active ingredient, excipient, or other additives to the drug product.The source of the impurity could be product or process related.
杂质:原料药或制剂中的任何成分,不是合成的化学或生物实体(定义为活性成分、辅料或制剂的其他添加剂)。杂质的来源可能与产品或工艺相关。
Intermediate: A material that is produced during a manufacturing process, which is not the final drug substance or the final drug product. Intermediates are identified by a manufacturer, who should establish and justify a control strategy to assure the intermediate’s stability within conditions of the manufacturing process. Bulk drug products are considered drug product intermediates.
中间产品:在生产过程中产生的物料,不是最终原料药或最终制剂。生产商应确定中间产品,建立并证明控制策略的合理性,以确保其在生产工艺条件下中间产品的稳定性。散装制剂被认为是制剂中间产品。
LED: Light-emitting diode
LED: 发光二极管
Long-term Testing: Stability studies under the recommended long-term storage condition for the re-test period or shelf life proposed (or approved) for labelling. Long-term testing results in real time data obtained at the long-term storage condition.
长期稳定性研究:在建议的长期条件下,针对说明书/标签建议(或批准)的复验期或有效期进行的稳定性研究。在长期放置条件下通过长期稳定性试验获得实时稳定性数据
Mass balance: For synthetic chemical entities, the process of adding together the assay value and levels of degradation products to see how closely these add up to 100% of the initial value, with due consideration of the margin of analytical error.
质量平衡:对于化学药物实体,在充分考虑了分析误差的情况下,将测定值和降解产物水平相加,以考察其是否接近初始值的100%。
Mean kinetic temperature: A single derived temperature that, if maintained over a defined period of time, affords the same thermal challenge to a drug substance or drug product as would be experienced over a range of both higher and lower temperatures for an equivalent defined period. The mean kinetic temperature is higher than the arithmetic mean temperature and takes into account the Arrhenius equation.
When establishing the mean kinetic temperature for a defined period, the formula of J. D. Haynes (28) can be used.
平均动力学温度:如果在相同特定的时期将原料药或制剂维持在该温度下,和同时经历较高和较低温度的情况相比,该推导温度可以对原料药或制剂提供相同的热挑战。平均动力学温度比算术平均温度高,其考虑了Arrhenius方程。在为一个特定的时期估计平均动力学温度时,可用J.D.Haynes(28)的公式来计算。
Model verification: The process of ensuring the model is implemented as intended. For example, confirmation that the modelled data for the initially proposed shelf life or re-test period are comparable to confirmatory experimental data.
模型确认:确保模型按预期实施的过程。比如,确认最初建议的有效期或复验期的模拟数据与验证性实验数据具有可比性。
Model validation: The process of determining the suitability of a model by challenging it with independent test data and comparing the results against predetermined performance criteria.
模型验证:通过用独立的测试数据对模型进行挑战,并将结果与预定的性能标准进行比较,来确定模型适用性的过程。
NMT: Not More Than Ongoing stability studies (also referred to as annual stability studies): Stability studies conducted under long-term storage conditions on an annual basis to ensure the consistency of stability related quality attributes at the approved storage conditions over the product lifecycle. These studies also allow for the monitoring of the stability characteristics and examine trends in the stability data to confirm the appropriate storage conditions relevant for the product and to confirm a re-test period or a shelf life.
NMT: 不得过持续稳定性研究(也称为年度稳定性研究):每年在长期条件下进行的稳定性研究,以确保在整个产品生命周期内,批准贮藏条件下的稳定性相关质量属性的一致性。这些研究还允许监测稳定性特征并检查稳定性数据的趋势,以确认与产品相关的适当贮藏条件,并确认复验期或有效期。
Open Dish Study: A study conducted without the protection of the immediate container, representing a worst-case scenario under controlled conditions.
敞口平皿研究:在没有直接容器保护的情况下进行的研究,代表受控条件下的最坏情况。
Pilot Scale Batch: A batch of an active pharmaceutical ingredient or finished pharmaceutical product manufactured by a procedure fully representative of and simulating that to be applied to a full production-scale batch. For example, for synthetics chemical entities in solid dosage forms, a pilot scale is generally, at a minimum, one-tenth that of a full production scale or 100 000 units, whichever is the larger, unless otherwise adequately justified. For biologics, the steps of upstream and downstream processing should be identical except for the scale of production.
中试规模批次:采用完全代表并模拟完整生产规模批次的工艺生产的一批活性药物成分或成品制剂。例如,对于固体剂型的化学药物实体,除非另有充分理由,否则中试规模通常至少为商业化生产规模的十分之一或至少生产100,000片或粒的规模,取较大者。对于生物制品,除生产规模外,上游和下游加工的步骤应相同。
PQS: Pharmaceutical Quality System
PQS: 药品质量体系
Primary Batch: A batch of a drug substance or drug product used in a primary stability study.
注册稳定性批次:用于注册的稳定性研究的原料药或制剂批次。
Primary Stability Studies: Stability studies conducted under the accelerated and long term (and, where applicable, intermediate) storage conditions undertaken on primary stability batches to establish a re-test period or a shelf life. Where appropriate, the primary stability studies may be conducted on non-production scale batches.
注册稳定性研究:在加速和长期条件(在适用情况下包括中间条件)下对注册稳定性批次进行的稳定性研究,以建立复验期或有效期。在适当的情况下,可在非生产规模批次中进行注册稳定性研究。
Prior Knowledge: Prior knowledge refers to existing knowledge and includes internal knowledge (e.g.,development and manufacturing experience), external knowledge (e.g., scientific and technical publications, including vendors’ data, literature and peer-reviewed publications), or the application of established scientific principles (e.g., chemistry, physics and engineering principles).
先验知识:先验知识是指现有的知识,包括内部知识(如开发和制造经验)、外部知识(如科学技术出版物,包括供应商的数据、文献以及经过同行评审的出版物),或对既定科学原理的应用(如化学、物理和工程原理)。
Production Batch: A batch of a drug substance or drug product manufactured at production scale using production equipment and process in the commercial production site as specified in the regulatory submission.
生产批次:在注册申报中规定的商业化生产场地,使用生产设备和工艺以生产规模生产的原料药或制剂批次。
Product lifecycle stability studies: Stability studies conducted under the accelerated, intermediate, or long-term storage conditions (as applicable) to support product lifecycle changes by assessing whether the change has an impact on any stability related quality attributes of the commercial drug substance or product under the labelled storage, handling and use conditions.
产品生命周期稳定性研究:在加速、中间条件或长期条件下(如适用)进行的稳定性研究,通过评估变更是否对市售原料药或产品在标示的贮藏、转运和使用条件下的任何稳定性相关质量属性产生影响,以支持产品生命周期变更。
RH: Relative Humidity
RH: 相对湿度
Re-test Date: The date after which samples of the drug substance should be examined to ensure that the material is still in compliance with the specification and thus suitable for use in the manufacture of a given drug product.
复验日期:在这一日期之后必须对原料药样品进行检验,以确保材料仍符合质量标准,并适用于生产规定的制剂。
Re-test Period: The re-test period is a period of time during which the drug substance is expected to remain within its specification and, therefore, can be used in manufacture of a given drug product, provided the drug substance has been stored under the defined conditions. After this period, a batch of drug substance can be re-tested for compliance with its specification and then used immediately for manufacture of drug product. A re-test period is normally applicable to synthetic drug substances and may be applicable to certain well-characterised biological drug substances.
复验期:在这段时间内,只要原料药贮藏于规定条件下就被认为其质量符合标准要求,可用于生产相应的制剂。此后,可重新检测一批原料药是否符合其质量标准,然后立即用于制剂生产。复验期通常适用于合成原料药,也可能适用于某些特性良好的生物原料药。
Semi-permeable Containers: Containers that allow the passage of solvent or gas, while preventing solute loss. Examples of semi- permeable containers include plastic bags and semi-rigid, low-density polyethylene (LDPE) pouches for large volume parenteral (LVPs), and LDPE ampoules, bottles and vials.
半渗透性容器:允许溶剂或气体透过,同时防止溶质损失的容器。半渗透性容器的示例包括用于大容量注射剂(LVP)的塑料袋和半硬质低密度聚乙烯(LDPE)袋,以及LDPE安瓿瓶、瓶子和小瓶。
Shelf life: The time period during which a drug substance or drug product is expected to remain within the approved shelf life specification, provided that it is stored under the conditions defined on the label.
有效期:在这段时间内,只要[原料药或]制剂在标签指定的条件下贮藏,就能符合经批准的有效期标准。
Significant Change for Synthetics: Significant change for a drug substance is defined as failure to meet its specification. In general, “significant change” for a drug product is defined as: (1) A 5% change in assay from its initial value; or failure to meet the acceptance criteria for potency when using biological or immunological procedures (e.g., for antibiotics); (2) Any degradation product exceeding its acceptance criterion; (3) Failure to meet the acceptance criteria for appearance, physical attributes and functionality test (e.g., colour, phase separation, re-suspendability, caking, hardness, dose delivery per actuation); however, some changes in physical attributes (e.g., softening of suppositories, melting of creams) may be expected under accelerated conditions; and, as appropriate for the dosage form; (4)Failure to meet the acceptance criterion for pH; (5) Failure to meet the specification for dissolution testing; or, (6) A 5% loss in water from its initial value for products stored in semi-permeable containers.
化学药物的显著变化:化学药物的显著变化定义为不符合其质量标准。一般而言,制剂的“显著变化”定义如下:(1)含量与初始值相比变化5%;或使用生物或免疫程序(如抗生素)时不符合效价的可接受标准;(2)任何降解产物超过其可接受标准;(3)不符合外观、物理属性和功能性测试的接受标准(如:颜色、相分离、再悬浮性、结块、硬度、每揿剂量);然而,在加速条件下可能会出现一些物理属性的变化(如栓剂变软、乳膏融化);视剂型而定。(4)未达到pH可接受标准;(5)溶出度检测不符合质量标准;或者(6)对于贮藏在半渗透性容器中的产品,其水分损失为初始值的5%。
Storage Condition Tolerances: The acceptable variations in temperature and relative humidity of storage facilities for formal stability studies.
贮藏条件公差:用于正式稳定性研究的贮藏设施的温度和相对湿度的可接受变化。
Stress Studies: Studies undertaken to assess the effect of stress conditions on the drug substance and/or drug product which can be divided into two categories:
影响因素稳定性研究:为评估影响因素条件对原料药和/或制剂的影响而进行的研究,可分为两类:
Studies conducted under stress conditions that are more severe than the accelerated conditions, but not necessarily intended to deliberately degrade the sample, which may be useful in gaining product knowledge and evaluating the effect of excursions outside the label storage conditions.
1) 在比加速条件更严格的影响因素条件下进行的研究,但不一定旨在故意降解样品,这可能有助于获得产品知识和评估偏离标签贮藏条件的影响。
Studies conducted under forced degradation conditions that are intended to deliberately degrade the sample (such as elevated temperature, humidity, pH, oxidation, agitation and light) and may be used to:investigate the potential degradation pathways; gain product knowledge; understand the intrinsic stability of drug substance; and used to develop and confirm stability-indicating nature of the analytical procedure.
2) 在强制降解条件下进行的研究,旨在故意降解样品(如:升高的温度、湿度、pH值、氧化、搅拌和光照),可用于:研究潜在的降解途径;获得产品知识;了解原料药的内在稳定性;并用于开发和确认指示分析方法性质的稳定性。
Supporting Data: Data, other than those from formal stability studies, that support the analytical procedures, the proposed re-test period or shelf life and the label storage statements. Such data include (1) stability data on early synthetic route batches of drug substance, small scale batches of materials, investigational formulations not proposed for marketing, related formulations and product presented in containers and closures other than those proposed for marketing; (2) information regarding test results on containers; and (3) other scientific rationales.
支持性数据:除了正式稳定性研究资料外,其他支持分析方法、建议的复验期或有效期以及标签贮藏说明的数据。这些数据资料包括(1)最初合成路线批次的原料药、小规模批次的材料、非拟上市试验剂型、相关剂型和包装产品(非拟上市产品)的稳定性数据;(2)有关容器试验结果的信息;(3)其他科学依据。
Supportive stability studies: Ancillary stability studies that are conducted (as applicable) to support the practical use of the product (including label claims) or a re-test period or a shelf life (e.g., photostability, in-use, short-term studies and studies to support excursions or modelling). Data to support short-term storage conditions, where relevant, may be provided as part of the primary stability studies.
支持性稳定性研究:为支持产品的实际使用(包括标签声明)或复验期/有效期而进行的稳定性研究(如适用)(如光稳定性、使用期间、短期研究和支持偏离或建模的研究)。支持短期条件的数据(如相关)可作为注册稳定性研究的一部分提供。
17 REFERENCES
17 参考文件
1. ICH Q2 Validation of Analytical Procedures/ ICH Q14 Analytical Procedure Development
2. ICH Q5E Comparability of Biotechnological/Biological Products Subject to Changes in their Manufacturing Process
3. ICH Q6A Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances)
4. ICH Q6B Specifications: Test Procedures and Acceptance Criteria for Biotechnology/Biological Products
5. ICH Q7 Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients
6. ICH Q8 Pharmaceutical Development
7. ICH Q9 Quality Risk Management
8. ICH Q10 Pharmaceutical Quality System
9. ICH Q11 Development and Manufacture of Drug Substances (Chemical Entities and
Biotechnological/Biological Entities
10. ICH Q12 Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle
Management
11. ICH Q13 Continuous Manufacturing of Drug Substances and Drug Products
12. ICH Quality Implementation Working Group Points to Consider (R2): Guide for ICH
Q8/Q9/Q10 Implementation
13. World Health Organisation. Technical Report Series 953, Annex 2, Appendix 1: Stability
testing of active pharmaceutical ingredients and finished pharmaceutical products: Stability
conditions for WHO Member States by Region (Updated March 2021).
14. WHO TRS 1010 - Annex 10: WHO guidelines on stability testing of active pharmaceutical ingredients and finished pharmaceutical products (2018).
15. Grimm W. Drugs Made in Germany. 1985;28;196-202 and 1986; 29:39-47
16. Grimm W. Drug Dev Ind Pharm. 1998;24(4):313-25
17.ISO/CIE International Organization for Standardization/International Commission on Illumination, International Standard 18909:2022,
18. Blümel A, et al. Journal of Pharmaceutical Sciences. 2023; 112(9):2332-2346,
19. Qiu F, Scrivens G. Accelerated predictive stability (APS): fundamentals and pharmaceutical industry practices. London, UK;2018
20. Murphy JR, Weisman D. Using random slopes for estimating shelf-life. Proceedings of the American Statistical Association of the Biopharmaceutical Section. 1990; p. 196-200
21. Carstensen JT. Stability and dating of solid dosage forms. In: Pharmaceutics of solids and solid dosage forms. Wiley-Interscience; 1977. p. 182-185
22. Ruberg SJ, Stegeman JW. Pooling data for stability studies: Testing the equality of batch
degradation slopes. Biometrics. 1991;47(4):1059-1069
23. Ruberg SJ, Hsu JC. Multiple comparison procedures for pooling batches in stability studies. Technometrics. 1992;34(4):465-472
24. Shao J, Chow SC. Statistical inference in stability analysis. Biometrics. 1994;50(3):753-763
25. Yoshioka S, Aso Y, Kojima S. Assessment of shelf-life equivalence of pharmaceutical products.Chem Pharm Bull (Tokyo). 1997;45(8):1482-4
26. Chen JJ, Ahn H, Tsong Y. Shelf-life estimation for multifactor stability studies. Drug Inf J.1997;31(3):573-587
27. Fairweather W, Lin TD, Kelly R. Regulatory, design, and analysis aspects of complex stability studies. J Pharm Sci. 1995;84(11):1322-1326
28. J. D. Haynes, J. Pharm. Sci., 60:927-929, (1971)
18 ANNEXES
18 附录
Annex 1 Reduced Stability Protocol Design
附录1-简化稳定性方案设计
A1-1 Introduction
A1-1 前言
This annex is intended to address recommendations on the application of reduced stability protocol designs conducted in accordance with principles outlined in the core guideline.
A reduced stability protocol design is one in which samples for every factor combination are not all tested at all time points.
本附录旨在根据核心指导原则中论述的原则,提出简化稳定性方案设计实施的应用建议。
简化稳定性方案设计是指并非在所有时间点对每个因素组合的样品都进行测试。
The reduced stability designs presented below may be proposed for any formal stability study protocol, i.e.,primary, commitment, ongoing (annual), product lifecycle. Implementation of some strategies requires a strong understanding of product stability performance and risks and may be more suitable for lifecycle applications or where prior knowledge may be leveraged.
以下列出的简化稳定性设计可以应用于任何正式稳定性研究方案中,如注册稳定性、稳定性承诺、持续(年度)稳定性研究或产品生命周期稳定性研究。一些策略的实施需要对产品稳定性表现和风险具有深入的理解,可能更适用于生命周期管理或能够应用先验知识的场景。
If a reduced protocol design is introduced after the original marketing authorisation, change management procedures should be followed (refer to ICH Q10) in accordance with regional requirements.
如果在初始上市许可后引入简化的方案设计,则应根据区域要求遵循变更管理程序(见ICHQ10)。
This annex provides guidance on bracketing and matrixing study designs and other science- and risk-based reduced stability design strategies. Specific principles are defined for situations in which reduced stability strategies can be applied. Sample designs are provided for illustrative purposes and should not be considered the only, or the most appropriate, designs in all cases.
本附录为括号法和矩阵法研究设计以及其他基于科学和风险的简化稳定性方案设计策略提供指导。为可以应用简化稳定性策略的情况,定义了具体的原则。样品设计仅用于说明目的,但不应被认为是在所有情况下的唯一或最恰当的设计。
A1-2 General Principles for Reduced Stability Designs
A1-2 简化稳定性设计的一般原则
Any reduced design should be able to meet the objective of the study with a defined and acceptable risk as compared to a full design. The potential risk associated with a reduced design should be considered (e.g.,establishing a shorter re-test period or shelf life than could be derived from a full design due to the reduced amount of data collected).
与完整设计相比,任何简化设计都应能够满足研究目标,且风险明确且可接受。应考虑与简化设计相关的潜在风险(比如,由于收集的数据量减少,确定比完整设计更短的复检期或有效期)。
Reduced designs can be applied to long-term stability studies for most types of drug products, although additional justification should be provided for complex products (e.g., a drug delivery system where there are many potential drug-device interactions, certain biological products). For the study of drug substances,matrixing is usually of limited utility and bracketing is generally not applicable; however, reduced time points and/or attribute testing could be justified where little or no degradation occurs. Additional reduced protocol designs are also discussed and may be most relevant when product and stability knowledge are high (e.g., to support post-approval changes; Refer to Section 15 – Stability Considerations for Commitments and Product Lifecycle Management).
简化设计可应用于大多数类型制剂的长期稳定性研究,但对于复杂产品(如有许多潜在药物-器械相互作用的给药系统、某些生物制品)应提供额外的依据。对于原料药的研究,矩阵法通常效用有限,括号法一般不适用;然而,在很少或没有降解发生的情况下,减少时间点和/或属性测试可以是合理的。还讨论了其他简化方案设计,当产品和稳定性知识掌握较充分时,这些设计可能最为相关(例如,支持上市后变更;见第15节“关于稳定性承诺和产品生命周期管理的稳定性考虑因素”)。
Whether a reduced design can be applied depends on a number of circumstances, as discussed in detail below. The use of any reduced design should be justified. In certain cases, the condition described in this annex is sufficient justification for use, while in other cases, additional justification should be provided.The type and level of justification in each of these cases will depend on the available supporting data and risk assessment.
简化设计能否应用取决于多种情况,下文将详细论述。任何简化设计的使用都应是合理的。在某些情况下,本附录中描述的条件足以证明其使用合理性,而在其他情况下,应提供额外的理由。在每种情况下,依据的类型和水平将取决于可用的支持性数据和风险评估。
The reduced designs discussed below are based on different principles. Therefore, careful consideration and scientific justification should precede the use of more than one reduced design principle together in one design.
下文讨论的简化设计基于不同的原则。因此,在一项设计中同时使用多个简化设计原则之前,应仔细考虑并进行科学论证。
If risks are identified during a reduced design study, a change to full testing or to a less reduced design may be implemented with an explanation of the drivers for the increase to the design. Proper adjustments should be made to the statistical analysis, where applicable, to account for the increase in sample size as a result of the change (26-27). Once the design is changed, full testing or less reduced testing should be carried out through the remaining time points of the stability study.
如果在简化设计研究期间发现风险,则可以变更为完整试验或简化程度较低的设计,并解释增加设计的驱动因素。在适用的情况下,应对统计分析进行适当调整,以应对由于变更而导致的样本量增加(26-27)。一旦设计发生变更,应在稳定性研究的剩余时间点进行全面检测或简化程度较低的检测。
A1-3 Reduced Design Approaches
A1-3 简化设计方法
A1-3.1 Bracketing
A1-3.1 括号法
Bracketing is design of a stability schedule such that only samples on the extremes of certain design factors,e.g., strength, package size, would be tested at all time points as in a full design. The design assumes that the stability of any intermediate levels is represented by the stability of the extremes tested. Bracketing can be applied to different container sizes or different fills in the same container closure system.
括号法是一种稳定性方案的设计方式,其特点是仅测试某些设计因素(如规格、包装尺寸)在极端条件下的样品,如同在完整设计中一样,在所有时间点进行测定。该方案假设,任何中间水平的稳定性都可以由所测试的极端情况的稳定性来代表。括号法可用于不同容器尺寸或同一容器密封系统中的不同装量。
The use of a bracketing design would not be considered appropriate if it cannot be demonstrated that the strengths or container sizes and/or fills selected for testing are indeed the extremes.
如果无法确证被选择受试的规格或容器尺寸和/或装量确实是处于极端情况,则认为使用括号法设计不合适。
A1-3.1.1 Design Factors
A1-3.1.1 设计因素
Design factors are variables (e.g., strength, container size and/or fill) to be evaluated in a study design for their effect on product stability.
设计因素是在研究设计中需要评估其对产品稳定性影响的变量(比如规格、容器尺寸和/或装量)。
A1-3.1.1.1 Strength
规格
Bracketing can be applied to studies with multiple strengths of identical or closely related formulations whose stability trends could be reasonably considered similar. Examples include but are not limited to (1)capsules of different strengths made with different fill plug sizes from the same powder blend, (2) tablets of different strengths manufactured by compressing varying amounts of a common blend, (3) liquid formulation of a biological of different concentration or fill volume, unless there are additional considerations for excluding some complex biologicals or live vaccines, (4) solutions and solid dosage forms for oral use of different strengths with formulations that differ only in minor excipients (e.g.,colourants, flavourings).
括号法可用于具有相同或相似处方的多规格制剂的研究,这些处方的稳定性趋势可以合理地被认为是相似的。示例包括但不限于(1)由相同粉末混合物的不同填充量制成的不同规格胶囊;(2)通过压制不同量的相同混合物而生产的不同规格的片剂;(3)不同浓度或装量的生物制品液体制剂,除非有额外考虑因素能够排除某些复杂生物制品或活疫苗;(4)处方仅在次要辅料(如着色剂、调味剂)上有所差异的不同规格的口服溶液剂和固体制剂其。
With justification and supporting data, bracketing can be applied to studies with multiple strengths where the relative amounts of drug substance and excipients change in a formulation.
在具备充分理由和支持性数据的情况下,括号法可适用于原辅料比例不同的多规格制剂的研究。
In cases where different excipients are used among strengths, bracketing generally should not be applied.
如果不同规格之间使用了不同的辅料,通常不应采用括号法。
A1-3.1.1.2 Container Closure Sizes and/or Fills
容器尺寸和/或装量
Bracketing can be applied to studies of the same container closure system where either container size or fill varies while the other remains constant. However, if a bracketing design is considered where both container size and fill vary, it should not be assumed that the largest and smallest containers represent the extremes of all container closure system configurations. Care should be taken to select the extremes by comparing the various characteristics of the container closure system that may affect product stability. Depending on the dosage form and container closure system, the following characteristics may be considered relevant:container wall thickness, closure geometry, surface area to volume ratio, headspace to volume ratio, water vapour permeation rate or oxygen permeation rate per dosage unit or unit fill volume, product contact coating, stopper or closure formulation and coating, as appropriate.
括号法可用于容器尺寸或装量不同,但其他保持不变的同种容器容器密封系统的研究。但是,如果采用括号法设计时容器大小和装量均不同,则不应假设最大和最小的容器代表了所有容器密封系统配置的极端情况。应注意通过比较可能影响产品稳定性的容器密封系统的各种特性来选择极端值。根据剂型和容器密封系统,以下特性可能是相关的:容器壁厚度、封闭几何形状、表面积与体积比、顶空与体积比、每单位剂量或单位装量体积的水蒸气渗透率或氧气渗透率、产品接触涂层、瓶塞或密封系统的配件和涂层等,视具体情况而定。
Bracketing can be applied to studies for the same container when the closure varies. Justification could include a discussion of the relative permeation rates of the bracketed container closure systems. Special consideration and justification may be required for drug products stored in semi-permeable containers (refer to Section 7.2.2 – Storage Conditions for Products Packaged in Semi-Permeable Containers).
括号法可用于同种容器不同闭塞物的研究。依据可包括对括号法包装密闭系统的相对渗透率的讨论。对于贮藏在半渗透性容器中的制剂,可能需要特殊考虑和说明(见第7.2.2节“半渗透性容器包装产品的贮藏条件”)。
A1-3.1.2 Design Considerations and Potential Risks
A1-3.1.2 设计考虑因素和潜在风险
Before a bracketing design is applied, its effect on the re-test period or shelf life estimation should be assessed. If the stability of the extremes is shown to be different, the intermediates should be considered no more stable than the least stable extreme (i.e., the shelf life for the intermediates should not exceed that for the least stable extreme).
If, after starting the studies, one of the extremes is no longer expected to be marketed, the study design can be maintained to support the bracketed intermediates.
在采用括号法设计前,应评估其对复检期或有效期的影响。如果极端条件的稳定性不同,则中间条件样品的稳定性不应超过最不稳定的极端条件(即中间条件样品的有效期不应超过最不稳定的极端条件的有效期)。
如果在研究工作开始后,其中一种极端情况预计不再上市销售,则可以维持该研究设计以支持括号法中间条件样品。
A1-3.1.3 Design Example
A1-3.1.3 设计示例
An example of a bracketing design is given in Table A1- 1. This example is based on a product available in three strengths and three container sizes. In this example, the 15 mL and 500 mL container sizes represent the extremes. The batches for each selected combination should be tested at each time point as in a full design. Note that the example below could represent multiple product types (synthetics and biologicals).
括号法设计示例见表A1-1。本示例基于一种有三种规格和三种容器尺寸的产品。在本示例中,15 mL和500 mL容器尺寸代表极端情况。同在完整设计中一样,每个选定组合的批次应在每个时间点进行试验。请注意,下面的例子可以代表多种产品类型(化学药物和生物制品)。
Table A1- 1: Example of a Bracketing Design
表A1-1 括号法设计示例
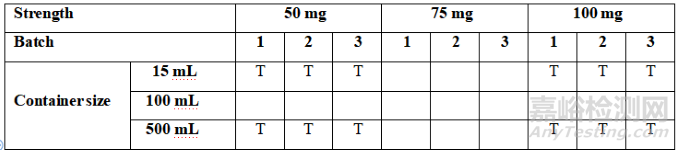
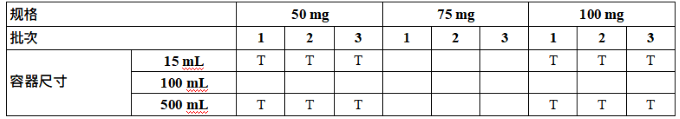
Key: T = Sample tested
注:T=试验样品
A1-3.2 Matrixing
A1-3.2 矩阵设计法
Matrixing is the design of a stability schedule such that a selected subset of the total number of possible samples for all factor combinations would be tested at a specified time point. At a subsequent time point,another subset of samples for all factor combinations is tested. The design assumes that the stability of each subset of samples tested represents the stability of all samples at a given timepoint. The differences in the samples for the same drug product should be identified, for example, covering different batches, different strengths, different sizes of the same container closure system and different container closure systems.
矩阵设计法是稳定性试验的设计方案,以便在指定的时间点对所有因素组合的可能样本总数中的选定子集进行测试。在随后的时间点,测试所有因素组合的另一个样本子集。该设计假设检测的每个样品子集的稳定性代表给定时间点所有样品的稳定性。应识别同一制剂样品的差异,如涵盖不同批次、不同规格、相同容器密封系统的不同尺寸以及不同的容器密封系统。
When a secondary packaging system contributes to the stability of the drug product, matrixing can be performed across the container closure systems (e.g., inclusion of a foil overwrap).
当次级容器密封系统影响制剂的稳定性时,可将该容器密封系统归入矩阵设计(例如,包括箔外包装)。
Each storage condition should be treated separately under its own matrixing design. Matrixing should not be performed across test attributes. However, alternative matrixing designs for different test attributes can be applied if justified.
每种贮藏条件应在其自身的矩阵设计下单独处理。矩阵设计不应跨试验属性进行。但是,如果合理,可以采用不同试验属性的替代矩阵设计。
A1-3.2.1 Design Factors
A1-3.2.1 设计因素
Matrixing designs can be applied to strengths with identical or closely related formulations. Examples include but are not limited to (1) capsules of different strengths made with different fill plug sizes from the same powder blend, (2) tablets of different strengths manufactured by compressing varying amounts of the same granulation and (3) oral solutions of different strengths with formulations that differ only in minor excipients (e.g., colourants or flavourings), (4) biological of different concentration and fill volume, (5)
biologicals of different concentration with different size container or pre-filled syringe size, (6) relative amounts of excipients (e.g., minor variation to the concentration of the filler).
矩阵设计可应用于具有相同或相似处方的不同规格制剂的研究。示例包括但不限于(1)由相同粉末混合物的不同填充量制成的不同规格的胶囊,(2)通过压制不同量的相同颗粒而制造的不同规格的片剂,(3)处方仅在次要辅料(例如着色剂或调味剂)上有所差异的不同规格的口服溶液,(4)不同浓度和填充体积的生物制品,(5)具有不同尺寸容器或预填充注射器尺寸的不同浓度的生物制品,(6)辅料的相对量(例如填充物浓度的微小变化)。
Justification should generally be based on supporting data. For example, to matrix across two different closures or container closure systems, supporting data could be supplied showing relative moisture vapour transmission rates or similar protection against light. Alternatively, supporting data could be supplied to show that the drug product is not affected by oxygen, moisture, or light.
论证通常应有支持性数据。例如,对于两种不同密封系统或容器密封系统的矩阵,可以提供显示相对水蒸气透过率或类似的避光保护措施的支持性数据。或者,可以提供表明制剂不受氧、水分或光影响的支持性数据。
Other factors for matrixing may be considered if justified, e.g., batches made by using the same process and equipment and container sizes and/or fills in the same container closure system.
如果合理,可以考虑其他矩阵设计因素,例如使用相同工艺和设备以及容器尺寸和/或相同的容器密封系统进行灌装的不同批次。
A1-3.2.2 Design Considerations
A1-3.2.2 设计考虑因素
A matrixing design should be balanced as far as possible so that each combination of factors is tested to the same extent over the intended duration of the study and through the last time point prior to submission.
矩阵设计应尽可能均衡,以便在预期的研究持续时间内和提交前的最后一个时间点对每种因素组合进行相同程度的试验。
However, due to the recommended full testing at certain time points, as discussed below, it may be difficult to achieve a complete balance in a design where time points are matrixed.
In a design where time points are matrixed, all selected factor combinations should be tested at the initial and final time points, while only certain fractions of the designated combinations should be tested at each intermediate time point. In addition, unless justified, data from at least three time points, including initial, should be available for each selected combination through the first 12 months of the study.
然而,如下所述,由于建议在某些时间点进行完整试验,因此在将时间点矩阵化的设计中可能难以实现完全均衡。在对时间点进行矩阵设计的方案中,所有选定的因素组合应在初始和最终时间点进行试验,而在每个中间时间点仅对指定组合中的某些部分进行试验。此外,除非在合理的情况下,否则在研究的前12个月,每个选定的组合应至少有三个时间点(包括起始时间点)的数据可用。
For matrixing at an accelerated storage condition, care should be taken to ensure testing occurs at a minimum of three time points, including initial and final, for each selected combination of factors. Thus, matrixing for accelerated studies may have limited application.
对于加速试验条件下的矩阵设计,应注意确保在至少三个时间点(包括初始和结束)对每个选定的因子组合进行试验。因此,加速试验的矩阵设计可能应用有限。
When a matrix on design factors is applied, if one strength or container size and/or fill is no longer intended for marketing, stability testing of that strength or container size and/or fill can be continued to support the other strengths or container sizes and/or fills in the design. Stability commitments in accordance with Section 15 – (Stability Considerations for Commitments and Product Lifecycle Management) should reflect the proposed commercial presentations.
当应用设计因素矩阵时,如果一种规格或容器尺寸和/或装量不再上市销售,则可以继续对该规格或容器尺寸和/或装量进行稳定性试验,以支持该设计方案中的其他规格或容器尺寸和/或装量。根据第15节“关于承诺和产品生命周期管理的稳定性考虑因素”做出的稳定性承诺应反映建议的商业展示。
A1-3.2.3 Design Examples
A1-3.2.3 设计示例
Examples of matrixing designs on time points for a product in two strengths (50 mg and 75 mg) are shown in Tables A1-2 and A1-3. The terms one-half reduction and one-third reduction refer to the reduction strategy initially applied to the full study design for timepoints excluding initial, 12-months and final. For example, a one-half reduction initially eliminates one in every two time points from the full study design and a one-third reduction initially removes one in every three. In the examples shown in Tables 2 and 3,the reductions are less than one-half and one-third due to the inclusion of full testing of all factor combinations at some time points.
两种规格(50 mg和75 mg)产品时间点的矩阵设计示例见表A1-2和表A1-3。术语“二分之一简化”和“三分之一简化”是指最初应用于完整研究设计的时间点(不包括初始、12个月和最终)的减化策略。例如,二分之一的简化最初会从完整研究设计中每两个时间点消除一个,三分之一的简化最初会每三个时间点消除一个。在表2和表3所示的示例中,由于在某些时间点纳入了所有因子组合的完整试验,因此简化量小于二分之一和三分之一。
Table A1- 2 Example One-Half Reduction Matrix Design on Time Points for a Product with Two Strengths
表A1-2 具有两种规格产品关于时间点的二分之一简化矩阵设计示例
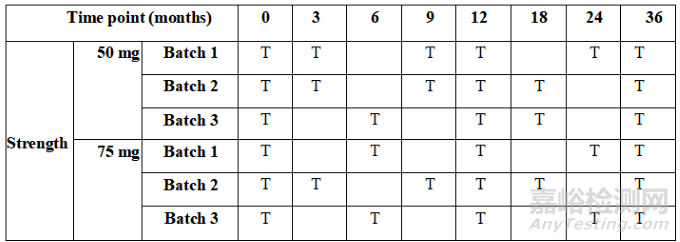
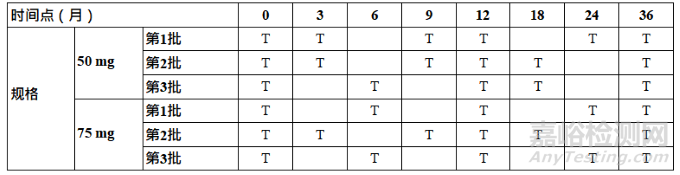
Key: T = Sample tested
注:T=试验样品
Table A1- 3 Example One-Third Reduction Matrix Design on Time Points for a Product with Two Strengths
表A1-3 具有两种规格的产品关于时间点的三分之一简化矩阵设计示例

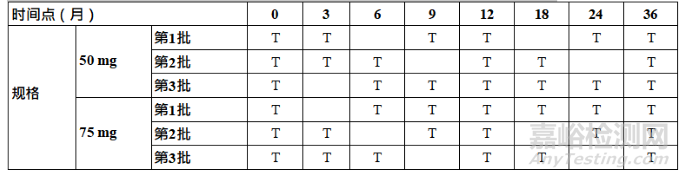
Key: T = Sample tested
Additional examples of matrixing designs for a product with three strengths (50 mg, 75 mg and 100 mg)and three container sizes (15 mL, 100 mL and 500 mL) are given in Tables A1-4 and A1-5. Table A1-4 shows a design with matrixing on time points only and Table 5 depicts a design with matrixing on time points and factors. In Table A1-4, all combinations of batch, strength and container size are tested, while in Table A1-5, certain combinations of batch, strength and container size are not tested.
注:T=试验样品
具有三种规格(50 mg、75 mg和100 mg)和三种容器尺寸(15 mL、100 mL和500 mL)的产品的矩阵设计的其他示例见表A1-4和表A1-5。表A1-4显示了仅对时间点进行矩阵设计的方案,表5描述了对时间点和因素进行矩阵设计的方案。在表A1-4中,对批次、规格和容器大小的所有组合进行了测试,而在表A1-5中,未对批次、规格和容器大小的某些组合进行测试。
Table A1- 4 Examples of Matrixing on Time Points for a Product with Three Strengths and Three Container Sizes
表A1-4 具有三种规格和三种容器尺寸的产品的时间点矩阵设计示例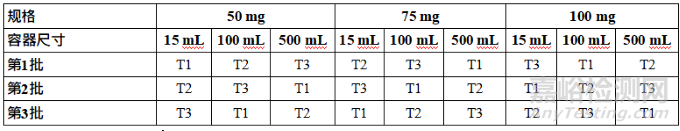
Table A1- 5 Examples of Matrixing on Time Points and Factors for a Product with Three Strengths and Three Container Sizes
表A1-5 具有三种规格和三种容器尺寸的产品的时间点和因素的矩阵设计示例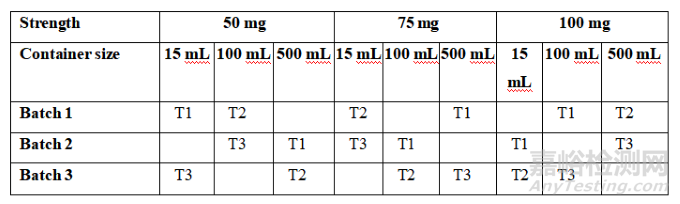
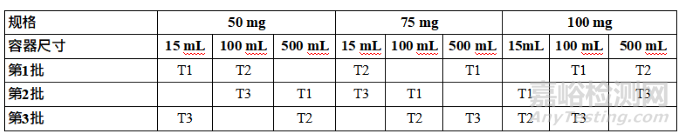
Key for Table A1- 4 and Table A1- 5:
表A1-4和表A1-5的说明:
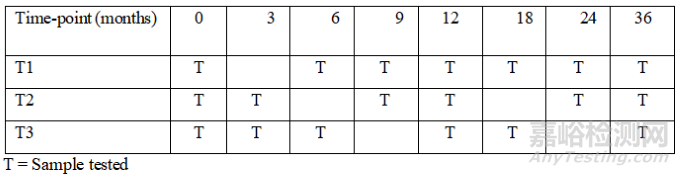
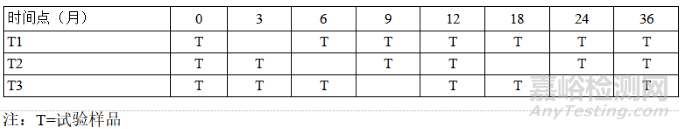
A1-3.2.4 Applicability and Degree of Reduction
A1-3.2.4 适用性和简化程度
The following, although not an exhaustive list, should be considered when a matrixing design is contemplated:
考虑矩阵设计时,应考虑以下因素(虽然不是详尽的清单):
knowledge of data variability
对数据变异性的理解
expected stability of the product
产品预期稳定性
availability of supporting data, including enhanced stability knowledge if available
支持性数据的有效性,包括进阶的稳定性知识(如有)
stability differences in the product within a factor or among factors
产品在一个因素内或因素间的稳定性差异
number of factor combinations in the study and/or
研究中的因素组合数量和/或
stability risk assessment, if performed.
稳定性风险评估(如进行)。
Data variability and product stability, as shown by supporting data, should be considered when a matrixing design is applied. If the supportive data show large variability, a matrixing design should not be applied.
应用矩阵设计时,应考虑支持性数据显示的数据变异性和产品稳定性。如果支持性数据显示变异性较大,则不应采用矩阵设计。
If a matrixing design is considered applicable, the degree of reduction that can be made from a full design depends on the number of factor combinations being evaluated. The more factors associated with a product and the more levels in each factor, the larger the degree of reduction that can be considered. However, any reduced design should have the ability to adequately predict the product shelf life.
如果认为矩阵设计适用,则完整设计的简化程度取决于所评价的因素组合的数量。与产品相关的因素越多,每个因素的层次越高,可以考虑的简化程度就越大。然而,任何简化设计都应该能够充分预测产品的有效期。
A1-3.2.5 Potential Risk
A1-3.2.5 潜在风险
Due to the reduced amount of data collected, a matrixing design on factors other than time points generally has less precision in shelf life estimation and yields a shorter shelf life than the corresponding full design.In addition, such a matrixing design may have insufficient power to detect certain main or interaction effects,thus leading to incorrect pooling of data from different design factors during shelf life estimation. If there is an excessive reduction in the number of factor combinations tested and data from the tested factor combinations cannot be pooled to establish a single shelf life, it may be impossible to estimate the shelf lives for the missing factor combinations. The risk may be mitigated through use of supportive stability data.
由于收集的数据量减少,对时间点以外的因素进行矩阵设计通常在有效期估计方面准确度较低,并且产生的有效期比相应的完整设计方案更短。此外,这种矩阵设计可能没有足够的能力来检测某些主要因素或因素间相互作用,从而导致在有效期评估期间,不正确地合并来自不同设计因素的数据。如果所测试的因素组合的数量简化过多,并且无法合并所测试的因素组合的数据以建立单一的有效期,则可能无法估计缺失的因子组合的有效期。可通过使用支持性稳定性数据来降低风险。
A study design that matrixes on time points only may be used to detect differences in rates of change among factors and to establish a reliable shelf life. This strategy assumes linearity and full testing of all other factor combinations at both the initial and final time points.
仅使用时间点矩阵的研究设计可用于检测因素之间变化率的差异,并确定可靠的有效期。该策略假设所有其他因子组合在初始和最终时间点,线性相关并进行完整试验。
A1-3.3 Knowledge and Risk Based Protocol Reductions
A1-3.3 基于知识和风险的方案简化
Additional reduced stability protocol designs that are different from bracketing and matrixing approaches may also be applied. Product knowledge and risk-based assessments are used to justify these stability strategies. If the knowledge- and risk-based reduced protocol is used to support a post-approval change, the risk assessment should also consider the potential impact of the change on the stability performance of the product. As discussed in ICH Q12, Chapter 9, there are numerous methods to assess the impact of a change in addition to long-term stability studies.
也可采用不同于括号法和矩阵法的其他简化稳定性方案设计。使用基于产品知识和风险评估来证明这些稳定性策略的合理性。如果使用基于知识和风险的简化方案来支持上市后变更,风险评估还应考虑变更对产品稳定性的潜在影响。如ICH Q12第9章所述,除了长期稳定性研究之外,还有多种方法可用于评估变更的影响。
A1-3.3.1 Design Factors
A1-3.3.1 设计因素
Where justified, a reduction may be applied to attributes, timepoints, samples and/or storage conditions.To apply these strategies, the applicant should present an understanding of what attributes are subject to change over the re-test period/shelf life and what conditions might impact their rate of change. This should be supported by data and/or product knowledge and used to conduct a risk assessment that justifies the proposed reductions.
在合理的情况下,可对属性、时间点、样品和/或贮藏条件进行简化。为了应用这些策略,申请人应说明哪些属性在复检期/有效期内会发生变化,以及哪些条件可能影响其变化率。这应提供数据和/或产品知识支持,并用于进行风险评估,以证明提出的简化策略是合理的。
A1-3.3.2 Design Considerations and Potential Risks
A1-3.3.2 设计考虑因素和潜在风险
Stability risk assessment tools should be developed throughout the product lifecycle in accordance with ICH Q9. The stability understanding used to assess risk may come from multiple sources, including stress testing, accelerated testing, formal stability studies and prior knowledge from product development, e.g.,on leachables and container closure integrity.
在整个产品生命周期中开发稳定性风险评估工具时应参考ICH Q9。用于评估风险的稳定性理解可能来自多个来源,包括强制降解稳定性试验、加速试验、正式稳定性研究和产品开发的先验知识,例如关于浸出物和容器密封完整性的先验知识。
Quality attributes that are considered low risk for stability testing are those that are unlikely to change on stability and are not critical to safety and efficacy of the product. An example of this is residual solvent content in a crystalline synthetic drug substance, since residual solvent content is assessed at release and will not increase over time and does not have the potential to impact other CQAs. With appropriate justification, these attributes may be removed from the stability protocol.
稳定性试验中被视为低风险的质量属性是指那些在稳定性期间不太可能发生变化且对产品的安全性和有效性不重要的质量属性。例如结晶来源的合成原料药中的残留溶剂含量,在放行时评估了残留溶剂含量,其不会随时间增加,也不会影响其他CQAs。在有适当依据的情况下,可从稳定性方案中删除这些属性。
Certain quality attributes may be removed when the attribute has the potential to change but has been demonstrated not to change over time or is monitored via other quality attributes and the change is established to not have a meaningful impact on quality, safety and efficacy through the re-test period or shelf life. However, to support a future change the impact on the stability of these quality attributes should be assessed and if necessary, reintroduced.
当某些质量属性有可能发生变化,但已证明不会随时间而变化,或者通过其他质量属性进行监测,并且确定该变化在整个复检期或有效期内不会对质量、安全性和有效性产生显著影响时,可以删除这些质量属性。但是,为了支持未来的变更,应评估这些质量属性对稳定性的影响,并在必要时重新引入。
A1-3.3.3 Design Strategies and Examples
A1-3.3.3 设计策略和示例
Descriptions of protocol reduction strategies and examples of instances where a reduced protocol approach may be applied with justification are provided below. These strategies may be applied to other situations as well when justified.
下面提供了方案简化策略的描述和可以应用简化方案方法的实例示例,并附有理由。在合理论证的情况下,这些策略也可应用于其他情况。
Reductions from the Primary Stability Protocol for Stability Commitments: Based on overall product knowledge, development data and/or results of the ongoing or completed primary stability study, the applicant may propose to remove attributes, storage conditions and/or timepoints for new protocols. This may be justified if the applicant:
稳定性承诺注册稳定性方案的简化:基于总体产品知识、开发数据和/或正在进行或已完成的注册稳定性研究的结果,申请人可提议删除新方案的属性、贮藏条件和/或时间点。如果申请人能够:
Demonstrates that the attribute is unchanging on stability, not clinically meaningful, not relevant to the assessment of re-test period or shelf life and not required for monitoring of the quality, safety and efficacy of the drug product after release and during its expected lifecycle.
证明该属性在稳定性上保持不变,无临床意义,与复检期或有效期的评估无关,并且无
需监测放行后和预期生命周期内制剂的质量、安全性和有效性。
Demonstrates how different storage conditions may impact stability and the worst-case storage conditions relevant to the drug substance or drug product are selected for evaluation.
证明不同的贮藏条件如何影响稳定性,并选择与原料药或制剂相关的最差贮藏条件进行评估。
Demonstrates that specific timepoints are not meaningful for assessment of trends.
证明特定时间点对趋势评估无意义。
Example 1 – Reduction from Primary Stability Study to the Commitment Study that Confirms Shelf Life for Synthetic Solid Oral Tablet.
示例1 - 从注册稳定性研究简化为确认合成固体口服片剂有效期的承诺研究。
Justification for the reduction from the primary protocol to the commitment protocol to confirm the shelf life (refer to Section 15 Stability Considerations for Commitments and Product Lifecycle Management)below may include historical data and accumulated knowledge supporting:
将注册方案简化为承诺方案以确认有效期的理由(见第15节“关于承诺和产品生命周期管理的稳定性考虑因素”)如下,可能包括历史数据和积累的知识支持:
lack of change to water activity and microbiological attributes, demonstration that trends are not significant justifying removal of the 9- and 18-month timepoints knowledge the product is stable when stored at 30 ºC/75 % RH and that this data may be used to represent storage at less strenuous room temperature conditions
水分活度和微生物属性无变化;证明趋势不显著,证明删除9个月和18个月时间点是合理的了解产品在30°C/75%RH条件下贮藏是稳定的,并且该数据可用于表示该产品可在较不苛刻的室温条件下贮藏
Table A1- 6 Example of a Protocol Design for Primary Stability Studies
Table A1- 6 注册稳定性研究的方案设计示例
A: Release Testing
A:放行检测
B: Appearance, Assay, Degradation Products, Dissolution, Water Content
B:外观、含量、降解产物、溶出度、水分
C: Appearance, Assay, Degradation Products, Dissolution, Water Content, Microbiological Testing
C:外观、含量、降解产物、溶出度、水分、微生物检测
Table A1- 7 Example of a Protocol Design for Commitment Stability Studies to Confirm Shelf Life
Table A1- 7 承诺稳定性研究确认有效期的方案设计示例
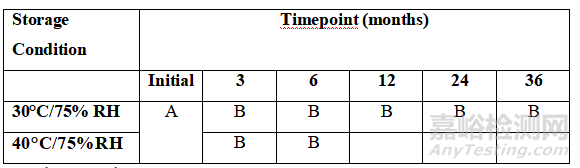

A: Release Testing
A:放行检测
B: Appearance, Assay, Degradation Products, Dissolution
B:外观、含量、降解产物、溶出度
Targeted Stability Designs:
目标稳定性设计:
Worst-Case Analysis Strategies: When stability characteristics for a product are well understood and a worst-case presentation is predictable, the applicant may design a stability strategy that evaluates the worst-case presentation with the conclusion that other presentations will demonstrate equivalent or better stability performance.
最差情况分析策略:当充分了解产品的稳定性特征,并且最差情况可预测时,申请人可设计一种稳定性策略,评估最差情况下的表现,并得出结论,其他展示将证明具有同等或更好的稳定性性能。
Example 2 - Different Drug Product Concentrations. If it is well-understood and predictable how the relative amounts of drug substance and excipients impact the stability profile for multiple concentrations,a worst-case approach could be proposed to support a reduction to samples. This approach may be justified where the concentration that provides the worst-case effect on stability is assessed. It is inferred based on product knowledge that if suitable stability is demonstrated for the worst-case concentration, the stability for other concentrations would be similar or improved.
示例2 - 不同制剂浓度。如果充分理解并可预测原料药和辅料的相对量如何影响多种浓度的稳定性特征,则可以提出一种最差情况方法来支持减少样品。如果评估了对稳定性产生最坏影响的浓度,则该方法可能是合理的。根据产品知识可以推断,如果在最坏情况的浓度下证明了适当的稳定性,则其他浓度下的稳定性将相似或有所提高。
Example 3 - Multiple Container Closure System Configurations and/or Fill Volume. If the characteristics of the product in different container sizes and/or with different fills are well understood and their impact on stability related quality attributes are predictable, then a worst-case approach could be proposed to support a reduction to samples. In this example, the configuration that presents the ‘worst-case’ for product stability is selected for the stability study. It is inferred based on product knowledge that if suitable stability is demonstrated for the worst-case configuration, the stability for other configurations would be similar or improved.
示例3 - 多种容器密封系统配置和/或装量。如果充分了解不同容器尺寸和/或不同装量的产品特性,并且其对稳定性相关质量属性的影响是可预测的,则可以提出一种最差情况方法来支持样品减少。在本示例中,选择呈现产品稳定性“最差情况”的配置进行稳定性研究。根据产品知识可以推断,如果证明最差情况下的配置具有适当的稳定性,则其他配置的稳定性将相似或有所提高。
A1-4 Data Evaluation for Reduced Study Designs
A1-4 简化研究设计的数据评估
The statistical procedures described in Section 13 - Data Evaluation can be applied to the analysis of stability data obtained from any reduced study design.
第13节 - 数据评估中描述的统计方法可应用于任何简化研究设计获得的稳定性数据的分析。
If a bracketing design is utilised, there is an assumption that the stability of the intermediate strengths or sizes/fills is represented by the stability at the extremes. If the statistical analysis indicates that the stability of the extreme strengths or sizes/fills is different, the intermediate strengths or sizes/fills should be considered no more stable than the least stable extreme. The statistical procedures suitable for multi-factor,full design study can be applied to the analysis of stability data obtained from a matrixing design study.The statistical analysis should clearly identify the procedure and assumptions used. The use of a matrixing design can result in an estimated shelf life shorter than that resulting from a full design.
如果采用括号法设计,则假设中等规格或尺寸/装量的稳定性由极限情况下的稳定性表示。
如果统计分析表明极限规格或尺寸/装量的稳定性不同,则应认为中等规格或尺寸/装量并不比最不稳定的极限更稳定。适用于多因素、完整设计研究的统计方法可应用于矩阵设计研究获得的稳定性数据的分析。统计分析应清楚地确定所使用的程序和假设。使用矩阵设计可能导致估计的有效期短于完整设计的有效期。
Where bracketing and matrixing are combined in one design or when an alternative reduced protocol is utilised, the same statistical principles may be applied.
当括号法和矩阵设计结合在一个设计中或使用替代简化方案时,可采用相同的统计原理。
Annex 2 Stability Modelling
附录2 稳定性建模
General information on selection of batches and minimum stability data at the time of submission and steps towards a comprehensive evaluation of available stability data are presented in Section 3 - Stability Protocol Design, Table 1 and Section 13 - Data Evaluation, respectively. When limited real-time data are available,Section 13.1 - General Considerations may be referenced for general considerations related to establishing an initial re-test or shelf life of drug substance or drug product using the decision tree for synthetics. While shelf life for biological products is generally established based on long-term stability data, enhanced stability modelling approaches could be considered for biological drug substances and drug products using the principles in section 2 of this Annex or using extrapolation principles (refer to Section 13.2.9-Extrapolation of Biologicals) for certain well-characterised biological drug substances with a well understood stability profile. This Annex provides additional and specific recommendations on statistical tools and models to support the use of extrapolation and enhanced stability modelling approaches.
关于批次选择和提交时的最低稳定性数据要求以及稳定性数据全面评估的步骤,请分别参见第3章《稳定性方案设计》表1和第13章《数据评估》。 当可用的实时数据有限时,可参考第13.1节《一般考量》,了解如何通过使用合成药物决策树确定原料药或制剂初始复检期或有效期。虽然生物制品的有效期通常根据长期稳定性数据确定,但对于某些具有良好稳定性特征的生物制品原液,可考虑采用本附录第2节中的原则对生物原料药和制剂进行增强稳定性建模或外推原则(参见第13.2.9节 - 生物制品的外推)。本附录进一步提供了支持外推法和增强建模的统计工具与模型的具体建议。
This Annex is structured in two parts, the first provides examples for the statistical tools and models commonly used to assess the data variability between batches for single factor and multi-factor, full design studies to establish re-test period or shelf life. The second part describes enhanced stability models for well-characterised molecules that may be based on empirical fit of stability data to kinetic functions or incorporating prior knowledge into data evaluation.
本附录分为两部分,第一部分提供了常用的统计工具和模型示例,适用于单因素及多因素完整设计研究,评估批次之间的数据变异性,以确定复检期或有效期。第二部分为增强稳定性模型,针对特性明确的分子,可基于稳定性数据与动力学函数的经验拟合,或将先验知识整合至数据评估。
As a general principle, the least complex statistical model that best describes the data is recommended to be used. Depending on the model and its context of use, the core study design elements that should be a part of any prospective stability modelling strategy include (1) defining the purpose of the model, (2) a description of the model, type of modelling (e.g., mechanistic or empirical) and its components, including specifying what is being estimated, tested for, or predicted, (3) identification of variables and appropriate statistical tools to achieve the stated study objectives, (4) sample size planning, (5) model development and fitting, including justification of the appropriateness of the input data (6) description, relevance and justification for use of product-specific prior knowledge and sources of prior knowledge, (7) model evaluation, including output data, limitations and assessing model robustness, (8) the quantitation and impact of uncertainty in any estimates or predictions providing adequate statistical assurance of any conclusions drawn (e.g., confidence, tolerance or prediction intervals) (9) model validation and verification with real-time data (10) plans for ongoing model monitoring and lifecycle considerations, as needed and (11) the risk management strategy if differences are observed between the predicted shelf life and actual shelf life based on confirmatory data. Consequently, its usage can be expected to be constrained by the modelling method, input or output data, conditions evaluated, etc., and should not be applied to conditions outside the model’s validated range, including different molecules, without a mechanistic understanding or robust scientific justification based on relevant prior knowledge. Refer to ICH Q8-10 Points to Consider for additional general principles related to model development, validation and verification. Models should be managed through a pharmaceutical quality system (PQS) after successful validation and verification.
作为一般原则,建议使用复杂度最低且能充分描述数据的统计模型。根据模型及其使用背景,应成为任何前瞻性稳定性建模策略一部分的核心研究设计要素包括:(1)定义模型的目的;(2)对模型、建模类型(如机械性或经验性)及其组成部分的描述,包括明确所估计、测试或预测的内容;(3)识别变量和适当的统计工具,以实现规定的研究目标;(4)样本量规划;(5)模型开发和拟合,包括输入数据适当性的论证;(6)产品特定先验知识和先验知识来源的说明、相关性和使用理由;(7)模型评价,包括输出数据、局限性和评估模型稳健性;(8)任何估计或预测中的不确定性的量化和影响,为得出的任何结论提供充分的统计保证(例如,置信度、公差或预测区间);(9)使用实时数据进行模型验证和确认;(10)根据需要制定持续模型监测和生命周期考虑计划;(11)制定风险管理策略(如果根据验证数据观察到预测有效期和实际有效期之间存在差异)。模型的使用会受到建模方法、输入或输出数据、评估条件等的限制。在没有基于相关先验知识的机械理解或可靠科学论证的情况下,不应应用于模型验证范围之外的条件,包括不同的分子。有关模型开发、验证和确认的其他一般原则,通用原则可参考ICH Q8-Q10中关于模型开发、验证和确认的要求。在该模型成功验证和确认后,应通过药品质量体系(PQS)管理模型进行管理。
A2-1 Statistical Evaluation of Stability Data from Single or Multi-factor Study Designs
A2-1 单因素或多因素研究设计的稳定性数据的统计评估
In this section of the Annex, data evaluation is discussed for (A) single factor and (B) multi-factor, full-design studies; where a single factor could be the batches used for a single product and multi-factors includes different fill volumes, concentrations, container dimensions etc, to set re-test period or shelf life when the stability protocol is not reduced by bracketing or matrixing (21). When data from non-primary batches are used, the representativeness of the process, container closure system and analytical procedure
should be justified, including the impact of any differences, in the context of the modelling strategy being proposed. Data from primary stability batches need to meet criteria outlined in Section 3 – Stability Protocol Design and elsewhere in this guideline. Useful references for the statistical approaches demonstrated in this guideline can be found in Section 17 – References (19, 25-27). Data evaluation for reduced study designs is described in Annex 1 (Reduced Stability Protocol Design) and Section 13 (Data Evaluation).
在附录的这一节中,讨论了(A)单因素和(B)多因素、完整设计研究的数据评估;其中单因素可以适用于单一产品的批次,多因素包括不同的装量、浓度、容器尺寸等,以便在稳定性方案未通过括号法或矩阵设计减少时设置复检期或有效期(21)。当使用非注册稳定性批次的数据时,需要在拟定的建模策略背景下证明工艺、容器密封系统和分析方法的代表性,包括任何差异的影响。注册稳定性批次的数据需符合第3节 - 稳定性方案设计和本指导原则其他章节中概述的标准。本指导原则中论证的统计方法的有用参考文献见第17节 - 参考文献(19、25-27)。简化研究设计的数据评估见附录1(简化稳定性方案设计)和第13节(数据评估)。
ICH Q1 CDE官方翻译中英文版3
A2-1.1 Evaluation of Variability for Stability Data in Single-factor, Full Design Studies
Using Linear Regression Models
A2-1.1 使用线性回归模型评估单因素、完整设计研究中稳定性数据的变异性
In general, the mathematical relationship between certain drug substance or drug product quantitative quality attributes and time is inferred to be linear as a reasonable approximation in a range of interest. The guideline (refer to Section 13 – Data Evaluation) describes how, for chemical synthetic entities, the available long-term stability data may be extrapolated to establish a shelf life using a decision tree approach.
Each primary, production and representative development batch in a formal stability protocol, stored under the long-term conditions, may be evaluated separately and the worst-case batch used to establish the re-test period or shelf life. Combining multiple batches is discussed in Annex 2, Section A2-1.2 - Linear Models Used to Assess Stability Profile and Section 13.2.2 - Combining Batches.
一般而言,推断某些原料药或制剂定量质量属性与时间之间的数学关系在相关范围内呈线性,为合理的近似值。本指导原则(参见第13节 - 数据评估)描述了如何使用决策树方法外推化学合成实体的可用长期稳定性数据,以确定有效期。正式稳定性方案中的每个注册稳定性批次、生产批次和代表性开发批次,如果在长期条件下贮藏,可单独进行评估,并将最坏情况下的批次用于确定复检期或有效期。附录2第A2-1.2节 - 用于评估稳定性特征的线性模型和第13.2.2节 - 合并批次中讨论了多个批次的合并。
Figure: A2- 1 shows the single batch (single-factor) regression line for assay of a synthetic chemical drug product with upper and lower acceptance criteria of 105 percent and 95 percent of label claim for assay, respectively. From 12 months of long-term data, a shelf life of 24 months can be proposed by extrapolation if no significant trends in accelerated and/or intermediate stability data. In this example, two-sided 95 percent confidence limits for the mean are calculated. The lower confidence limit intersects the lower acceptance criterion at 30 months, while the upper confidence limit does not intersect with the upper acceptance criterion until later. Therefore, the proposed shelf life of 24 months can be supported by the statistical analysis of the assay. A similar approach may be used for an attribute, such as an impurity, that increases over time and has a one-sided upper 95% confidence limit intersecting the attribute specification and support the target shelf life (: Shelf Life Estimation with Upper and Lower Acceptance Criteria
图A2-1显示了合成化学药品含量的单批次(单因素)回归线,其可接受标准上限和下限分别为含量标示量的105%和95%。根据12个月的长期稳定性数据,如果加速和/或中期稳定性数据无显著趋势,则可通过外推法拟定有效期为24个月。在本例中,计算平均值的双侧95%置信限。置信下限在30个月时与可接受标准下限相交,而置信上限在之后才与可接受标准上限相交。因此,该试验的统计分析可支持拟定的有效期为24个月。对于随时间推移而增加且单侧95%置信上限与属性质量标准相交并支持目标有效期(如杂质),可使用类似方法进行评估。

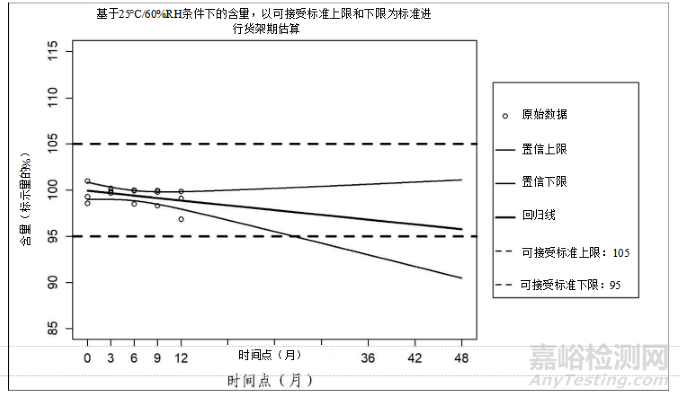
Figure: A2- 2). When the above approach is used, the mean value of the quantitative attribute (e.g., assay, degradation products) can be expected to remain within the acceptance criteria through the end of the re- test period or shelf life at a confidence level of 95 percent.
(根据图A2-2的上、下限可接受标准下的有效期估算)。当使用上述方法时,定量属性(例如含量、降解产物)的平均值在95%的置信度水平下预计在复检期或有效期结束前保持在可接受标准范围内。
Figure: A2- 1: Shelf Life Estimation with Upper and Lower Acceptance Criteria
图A2-1 上、下限可接受标准下的有效期估算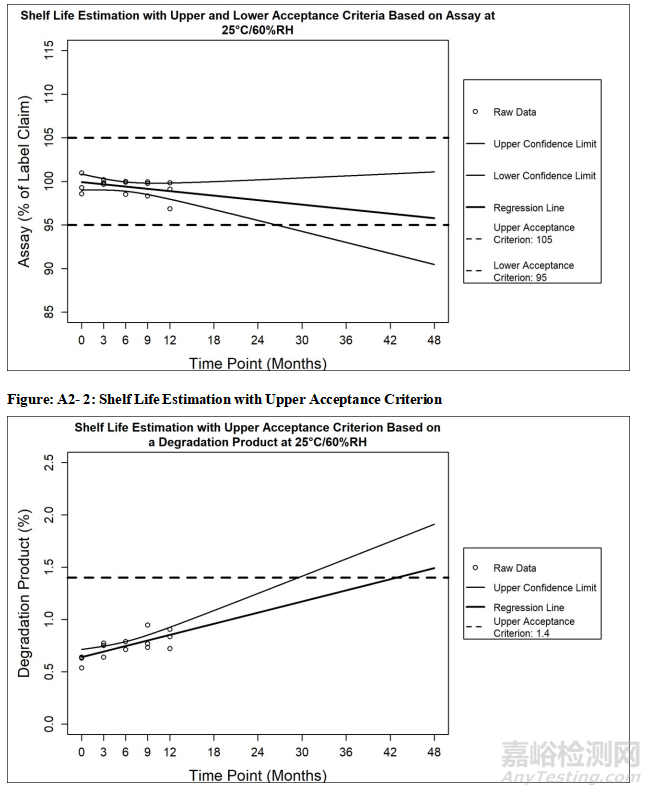
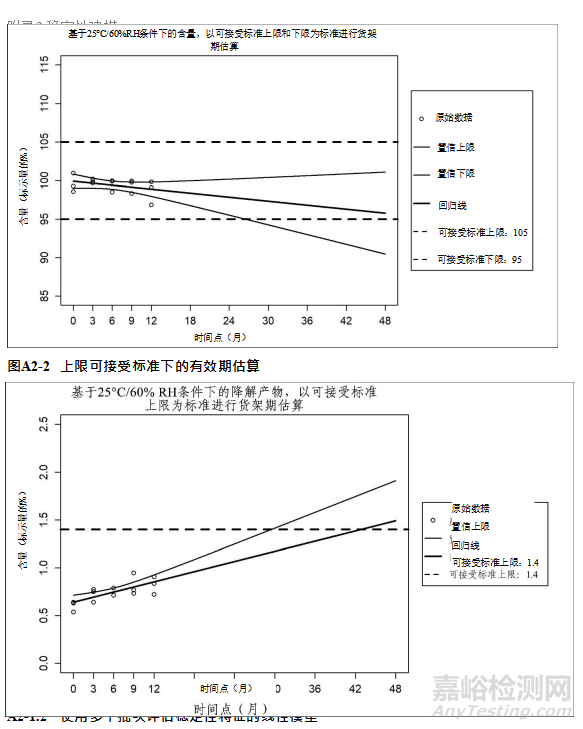
A2-1.2 Linear Models to Assess Stability Profile Using Multiple Batches
When stability data for more than a single batch are available, the data evaluation may use a linear model to evaluate the attribute stability profile at stated storage conditions and either establish or support a re-test period or shelf life. A linear model (Analysis of Covariance (ANCOVA), fixed effects or mixed effects model) may be applied to stability data, in which, the aim is to generate confidence bounds (or tolerance intervals for mixed effect models) and establish the maximum re-test period or shelf life that may be claimed.
The accuracy and precision of the analysis is determined by the number of batches suitable for the analysis, confidence in the uniformity of data, and the number of data points within each time-course study.
Applicants are advised that there is inherent risk of inaccurate representation of the stability profiles of manufactured batches dependent on the number of batches used and that batch numbers should be a consideration for study design. The minimum data set is discussed in the guideline (refer to Section 3 –Stability Protocol Design). A confidence interval based approach may be applied to evaluate shelf life when long-term data through shelf life are available (20).
当多个批次的稳定性数据可用时,可使用线性模型进行数据评估,以评估规定贮藏条件下属性的稳定性曲线,并建立或支持复检期或有效期。线性模型(协方差分析(ANCOVA)、固定效应或混合效应模型)可应用于稳定性数据,其目的是生成置信限(或混合效应模型的容许区间),并建立可能声称的最大复检期或有效期。分析的准确度和精密度由适用于分析的批次数量、数据均匀性的置信度以及每个时程研究中的数据点数量决定。申请人应注意,根据所用批次的数量,生产批次的稳定性特征存在不准确表示的固有风险,并且批号应是研究设计的考虑因素。指导原则中讨论了最小数据集(参见第3节 - 稳定性方案设计)。当有效期内的长期数据可用时,可采用基于置信区间的方法来评估有效期(20)。
Two model types are outlined below for the linear regression evaluation of stability data to establish re-test period or shelf life, fixed effects and mixed effects models. The models transform according to whether the batches are considered as a fixed (refer to Annex 2-Stability Modelling, Section 1.2.1 – Fixed Effects Model) or random variable (refer to Annex 2, Section 1.2.2 – Mixed Effects Model) and whether the variables are fixed or random. The choice of model generally depends on the number of batches used for the evaluation.
下面概述了两种模型类型,用于稳定性数据的线性回归评估,以确定复检期或有效期、固定效应模型和混合效应模型。根据批次是被视为固定变量(参见附录2 - 稳定性建模中第1.2.1节 - 固定效应模型)还是随机变量(参见附录2第1.2.2节 - 混合效应模型)以及变量是固定变量还是随机变量,对模型进行转换。模型的选择通常取决于用于评估的批次数量。
A2-1.2-1 Fixed Effects Model
固定效应模型
A Fixed Effects Model may be chosen when limited batches are available, e.g., three primary stability batches. The ANCOVA Fixed Effects Model expresses the attribute value at each timepoint and each batch as a function of the average y-intercept and average slope with their respective variability across the batches.
The level of significance for similarity between batches for intercept and slope should be proportionate to the number of batches used in the analysis, where a higher number of batches leads to lower significance level. When only 3 batches are available representative of the production batches, the model may consider batch as a fixed effect rather than as a random variable, with a selected significance level (p-value) for intercept and slope of 0.25. From regression lines, 95% confidence bounds for attributes may be one-sided or two-sided, depending on their acceptance criteria and if the attribute is known to be increasing or
decreasing, e.g., a purity attribute typically has a one-sided acceptance criterion, whereas potency, for a biological drug substance or drug product, typically has two-sided acceptance criteria. Increasing the significance level for one-sided confidence intervals may be appropriate.
当可用批次数量有限时,例如三个注册稳定性批次,可以选择固定效应模型。ANCOVA固定效应模型将每个时间点和每个批次的属性值表示为平均y截距和平均斜率及其各自在批次间的变异性的函数。批次之间截距和斜率相似性的显著性水平应与分析中使用的批次数量成比例,批次数量越多,显著性水平越低。当只有3个批次代表生产批次时,模型可以将批次视为固定效应而非随机变量,截距和斜率的选定显著性水平(p值)为0.25。根据回归线,属性的95%置信区间可能是单侧或双侧,这取决于其可接受标准,以及属性是否已知是增加或减少,例如,纯度属性通常具有单侧可接受标准,而生物制品原液或制剂的效价通常具有双侧可接受标准。增加单侧置信区间的显著性水平可能是合适的。
The possible models after sequentially evaluating the significance of slope variability and intercept variability between batches are indicated in Figure 3. There is no option for Different Slopes, Common Intercept because it is not realistic from a practical perspective to have all batches start from the same initial value at time (t)=0, but then have different slopes. When there is a distribution for the attribute at release (time zero), owing to lot-to-lot and assay variability, the model allows for different intercepts between batches.
依次评估批次间斜率变异性和截距变异性的显著性后,可能的模型如图3所示。无不同斜率、共同截距的选项,因为从实际角度来看,在时间(t)=0时使所有批次从相同的初始值开始,但随后具有不同的斜率并不现实。当放行时(零时间点)的属性存在分布时,由于批间和测定变异性,模型允许批次之间的不同截距。
Scenario A: When the statistical analysis demonstrated no statistically significant differences among slopes and no statistically significant differences among y-intercepts (p-values > 0.25), the batch term is dropped from the model and a common slope/common intercept model is fit to the data, which can be recognised as a simple linear regression model supporting, in this example, a 24 months shelf life based on the confidence bound crossing the shelf life specification acceptance limit at or after the proposed shelf life.
场景A:当统计分析显示斜率之间无统计学显著性差异,y截距之间无统计学显著性差异(p值 > 0.25)时,将批次项从模型中删除,并将共同斜率/共同截距模型拟合至数据。在本例中,这可被视为一种简单的线性回归模型,基于在拟定有效期或之后超过有效期质量标准可接受标准的置信限,最终确认有效期为24个月。
Scenario B: For attributes where the differences between the slopes were not statistically significant (p-value > 0.25), but differences between the y-intercepts were statistically significant (p-value < 0.25), the common slope/different intercepts model was used as the final model. The worst-case batch was identified as described in Figure 3 (batch #3). The shelf life is met if the worst-case batch’s confidence bound crosses the shelf life specification acceptance limit at or after the proposed shelf life (e.g., after 18 months).
场景B:对于斜率之间的差异无统计学意义(p值>0.25),但y截距之间的差异有统计学意义(p值<0.25)的属性,使用共同斜率/不同截距模型作为最终模型。最坏情况批次如图3所示(批次#3)。如果最坏情况批次的置信限在拟定的有效期或之后(例如,18个月后)超过有效期质量标准可接受标准,则符合有效期要求。
Scenario C: For attributes where the differences between slopes were statistically significant (p-value<0.25), the different slopes/different intercepts model was used as the final model. The worst-case batch is the one whose confidence bound yields the earliest intersection with shelf life specification acceptance limit (batch #1). The shelf life is claimed if the worst-case batch’s 95% confidence bound crosses the shelf life specification acceptance limit at or after the proposed shelf life (e.g., after 24 months).
场景C:对于斜率之间的差异具有统计学意义(p值<0.25)的属性,使用不同斜率/不同截距模型作为最终模型。最差情况批次是指其置信限与有效期质量标准可接受标准最早相交的批次(批次#1)。如果最坏情况批次的95%置信区间在拟定的有效期或之后(例如,24个月后)超过有效期质量标准可接受标准,则声明该有效期。
Figure A2- 3: Potential Final Models After Evaluating Slope and Intercept
图A2-3 评估斜率和截距后的潜在最终模型
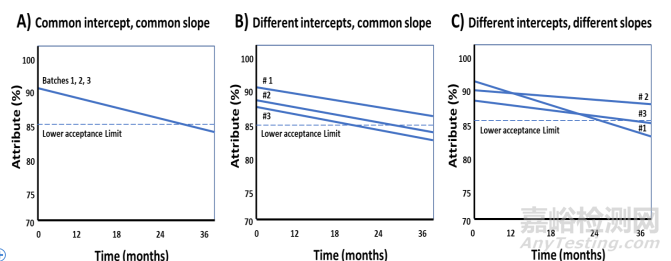
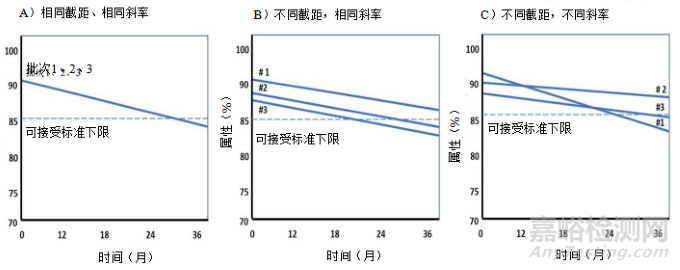
The final models per attribute may then be used to predict the mean attribute values and the 95% confidence bound(s), to establish the re-test period or shelf life as no more than the point of intersection of the appropriate upper or lower confidence bound with the attribute specification.
然后,每个属性的最终模型可用于预测平均属性值和95%置信限,以确定复检期或有效期不超过相应置信上限或下限与属性质量标准的交点。
Mixed Effects Model
混合效应模型
A mixed effects model may be chosen when five or more batches are available for statistical evaluation so that batch can be treated as a random variable. Batches, in addition to those defined as the primary stability batches, would be deemed as sufficiently representative of the primary batches and future production batches through analytical comparability with differences concluded to not impact the stability profile of the drug substance or drug product. A mixed effects model is recommended when there is risk to batch uniformity (i.e., greater risk of batch to batch variability). If the variance components for the random slope and intercept terms are estimated to be or close to zero (0), applying the fixed effect model can be more appropriate.
当有五个或更多批次可用于统计评估时,可以选择混合效应模型,以便将批次视为随机变量。通过分析可比性,认为除定义为注册稳定性批次的批次外,其他批次也足以代表主要批次和 未来生产批次,得出结论:差异不会影响原料药或制剂的稳定性特征。当存在批次均匀性风险(即批次间变异性的风险较大)时,建议使用混合效应模型。如果随机斜率和截距项的方差分量估计为零或接近于零(0),则应用固定效应模型可能更合适。
The mixed effects model reflects the expectation of random variation among the batches in terms of initial levels and trends over time (i.e., intercepts and slopes for a linear model), and hence the true shelf life is unique to each batch. The larger number of batches provides greater assurance that the inferred stability profile is representative of future batches manufactured using the same process. A tolerance interval-based approach using the linear mixed effects model may be applied to determine an extended shelf life beyond the period covered by long-term data. For instance, the shelf life of the product is determined as the (latest) timepoint where the (95%) the lower confidence limit of the 5th percentile (or the lower limit of the 95%/90% tolerance interval – first percentage refers to population covered, second confidence level) of the CQA is above acceptance limits. Corresponding tolerance interval-based approaches may be used to extrapolate an extended shelf life beyond the period covered by long-term data from the linear mixed effects model.
混合效应模型反映了在初始水平和随时间变化的趋势(即线性模型的截距和斜率)方面对批次间随机变化的预期,因此每个批次的真实有效期是唯一的。批次数量越多,越能确保推断的稳定性特征对未来采用相同工艺生产的批次具有代表性。使用线性混合效应模型的基于容许区间的方法可用于确定超出长期试验数据覆盖期限的延长有效期。例如,产品的有效期确定为CQA第5百分位数(或95%/90%容许区间的下限-第一个百分比是指覆盖的群体,第二个置信水平)的(95%)置信下限高于可接受标准的(最晚)时间点。相应的基于容许区间的方法可用于推断超出线性混合效应模型长期试验数据覆盖期限的延长有效期。
A2-2 Enhanced Stability Modelling
A2-2 增强稳定性建模
This section provides scientific and regulatory considerations for enhanced stability model development,qualification and maintenance over product lifecycle for the purpose of supporting a re-test period or shelf life. Guidance is provided for stability models that may be applied to well-understood drug substances or drug products that have been extensively characterised, including the identification of their relevant degradation pathways. When enhanced stability modelling is used, applicants are encouraged to consult with regulatory authorities to understand submission expectations.
本节提供了产品生命周期内增强稳定性模型开发、确认和维护的科学和监管考虑因素,以支持复检期或有效期。为稳定性模型提供了指导,这些模型可应用于已广泛表征的已知原料药或制剂,包括确定其相关降解途径。当使用增强稳定性建模时,鼓励申请人咨询监管机构,以了解提交预期。
Focus is placed on the design and data evaluation of enhanced stability models that can evaluate and extrapolate linear and non-linear quality attribute changes over time and includes the use of prior knowledge.
Linear regression for the extrapolation of stability data and the use of stability data from different batches are discussed in the core guideline (refer to Section 13 – Data Evaluation) and Section 1 of this Annex(refer to Section A2-1 Statistical Evaluation of Stability Data from Single or Multi-factor Study Designs).
重点是强化稳定性模型的设计和数据评估,该模型可以评估和外推线性和非线性质量属性随时间的变化,并包括使用先验知识。核心指导原则(参见第13节 - 数据评估)和本附录第1节(参见第A2-1节单因素或多因素研究设计的稳定性数据的统计评估)讨论了用于外推稳定性数据的线性回归和使用不同批次的稳定性数据。
A2-2.1 General Principles of Enhanced Stability Modelling
A2-2.1 增强稳定性建模的一般原则
The principles described in the ICH Points to Consider guide to implement ICH Q8/Q9/Q10, apply to stability models that are used to extrapolate re-test period or shelf life. These concepts are expanded in the subsequent sections of this annex. A stability model used to set commercial re-test period or shelf life would be considered a High-Impact Model in accordance with the elements for consideration in model validation,verification and documentation and would be of higher risk, than, for example models used during development studies.
ICH Q8/Q9/Q10实施指导原则中描述的原则适用于外推复检期或有效期的稳定性模型。这些概念在本附录的后续章节中进行了扩展。根据模型验证、确认和记录中考虑的要素,用于设定商业复检期或有效期的稳定性模型将被视为高影响模型,并且与开发研究中使用的模型相比,风险更高。
There are many types of stability models available or currently under development and, correspondingly, the tools to evaluate data from such stability models. This annex covers general principles of currently known kinetic, thermo-kinetic and mechanistic models as well as in silico or de novo computational methods that simulate known attribute stability profiles. This annex does not attempt to be comprehensive in describing all possible stability models or means of model data evaluation that could be considered acceptable when justified. Stability models may be empirical in nature by fitting the available stability data and known variables to derived mathematical relationships that describe how the quality attribute stability profile changes over time and measured under defined conditions. While enhanced stability models may be used to predict the stability profile at submission, these models should be considered as part of a comprehensive stability program and are not intended to replace long-term stability studies.
有许多类型的稳定性模型可用或目前正在开发中,相应地,还有评估此类稳定性模型数据的工具。本附录涵盖了目前已知的动力学、热动力学和机械模型的一般原理,以及模拟已知属性稳定性特征的计算机模拟或全新计算方法。本附录不试图全面描述所有可能的稳定性模型或合理时可接受的模型数据评估方法。稳定性模型本质上可以是经验性的,通过将可用的稳定性数据和已知变量拟合到推导的数学关系中,这些数学关系描述了质量属性稳定性特征如何随时间变化,并在规定的条件下进行测量。虽然增强稳定性模型可用于预测提交时的稳定性特征,但这些模型应被视为全面的综合稳定性研究计划的一部分,并不旨在取代长期稳定性研究。
An enhanced level of understanding for a drug substance or drug product under development (ICH Q8),which may encompass prior knowledge from drug substance or drug product development studies and information from structurally and functionally related molecules, referred to as “analogous molecules”,enables the use of stability models. See Sections 2 (Development Studies under Stress and Forced Conditions) and 3 (Stability Protocol Design) for considerations for prior knowledge. The sum of knowledge of the available stability data including confirmatory data, could support a quantitative prediction model.
There are many situations when a stability model may be applicable, including: setting the re-test period or shelf life and assessing the impact of storage condition excursions or manufacturing changes. A stability model may be applied during drug substance or drug product development, for an initial regulatory submission or as a post-approval, lifecycle management activity. The purpose of the model and the specific context of its use should be clearly stated.
加强对正在开发的原料药或制剂(ICH Q8)的理解,包括来自原料药或制剂开发研究的先验知识以及结构和功能相关分子(称为“类似分子”)的信息,可以使用稳定性模型。有关先验知识的注意事项,请参见第2节(强力条件和强制降解条件下的开发研究)和第3节(稳定性方案设计)。包括确认数据在内的现有的稳定性数据的知识总和,可以支持定量预测模型的建立。
稳定性模型可能适用于多种情况,包括:设定复检期或有效期以及评估贮藏条件偏离或生产变更的影响。稳定性模型可用于原料药或制剂开发、初始监管提交或批准后的生命周期管理活动。应明确说明模型的目的及其使用的具体背景。
A2-2.2 Model Development
A2-2.2 模型开发
A2-2.2.1 Choice of Model Type
A2-2.2.1 模型类型的选择
Certain types of stability model are built using data obtained at elevated conditions of temperature and/or humidity. The experimental accelerated conditions may be a selected set of defined parameters that may or may not overlap with the formal accelerated and stressed storage conditions as described in Section 7 - Storage Conditions.
某些类型的稳定性模型是使用在升高的温度和/或湿度条件下获得的数据建立的。实验加速条件可以是一组选定的定义参数,这些参数可能与第7节“贮藏条件”中描述的正式加速和强制降解贮藏条件重叠,也可能不重叠。
Depending on the underlying principles of the proposed stability modelling methodologies, the product type under consideration and the specific purpose of the model, certain model types may be more appropriate than others. The choice of model could depend on:
根据拟议稳定性建模方法的基本原则、所考虑的产品类型和模型的具体目的,某些模型类型可能比其他模型更合适。模型的选择取决于:
the intended context of use for the model,
模型的预期使用环境,
fit of stability data at the recommended storage condition to a kinetic formula,
推荐贮藏条件下的稳定性数据与动力学公式的拟合程度,
thermo-kinetic reactions with the fit of Arrhenius equation or its derivatives to stability data at accelerated temperatures,
用Arrhenius方程或其导数拟合加速温度下稳定性数据的热动力学反应,
the access to relevant prior knowledge,
获取相关先验知识,
the nature of the shelf life limiting attributes, their criticality ranking, impact on the stability profile and known correlations with structure or function of the molecule.
有效期限制属性的性质、其关键性等级、对稳定性特征的影响以及与分子结构或功能的已知相关性。
The appropriateness of the selected model for its intended use should be briefly described and justified in the specific context of its proposed use. The model should be described in sufficient detail to understand how it was developed and how the model is used to provide accurate prediction or inference of a quality attribute stability profile.
应简要描述所选模型对其预期用途的适用性,并在其拟议用途的具体背景下证明其合理性。应充分详细地描述该模型,以了解其开发方式以及如何使用该模型准确预测或推断质量属性稳定性特征。
A biological drug substance or drug product may be less amenable to modelling by the humidity modified Arrhenius equation using accelerated condition data, whereas the temperature/humidity-dependent kinetics for a solid synthetic chemical drug substance or drug product may obey the humidity modified Arrhenius equation for the shelf life limiting attributes. In addition, a model, on a case-by-case basis, may not be appropriate for physical attribute changes.
生物制品原液或制剂可能不太适合使用加速条件数据的湿度修正Arrhenius方程进行建模,而固体合成化学原料药或制剂的温度/湿度依赖性动力学可能遵循有效期限制属性的湿度修正Arrhenius方程。此外,在特定情况下,某些模型可能不适合用于物理属性变化的建模。
Enhanced stability models can fall under two broad classes: (1) those that utilise only the product-specific representative batch stability data (long-term and/or accelerated) and (2) those that additionally utilise prior knowledge from analogous-molecules combined with the product-specific information. Prior knowledge may be incorporated into the stability model evaluation in different ways, for example, to establish an acceptable range for the attribute stability profile, or by using Bayesian statistics.
增强稳定性模型可分为两大类:(1)仅使用产品特定的代表性批次稳定性数据(长期和/或加速)的模型,以及(2)额外利用类似分子先验知识与产品特定信息的模型。可以通过不同的方式将先验知识纳入稳定性模型评估中,例如,建立属性稳定性特征的可接受范围,或者通过使用贝叶斯统计。
It is recognised that novel model types are likely to emerge in the future (e.g., use of Artificial Intelligence Machine Learning, AI-ML). The principles outlined in this Annex should be generally applicable when developing a novel stability model, though other considerations regarding data requirements may also apply.
未来也可能会出现新的模型类型(例如,使用人工智能机器学习,AI-ML)。本附录中概述的原则在开发新的稳定性模型时应普遍适用,尽管有关数据要求的其他考虑因素也可能适用。
Early engagement with regulators is recommended in such instances.
在这种情况下,建议尽早与监管机构沟通。
2.2.1 Selection of Critical Quality Attributes for Stability Modelling
2.2.1 稳定性建模的关键质量属性的选择
Those attributes selected for modelling should be chosen according to the purpose of the model and the available stability knowledge. Those attributes not selected for modelling should be justified. The selection of CQAs for modelling follows the same principles described in the core guideline for protocol design and adapted to the purpose of stability model development. The selection of stability-indicating CQAs used in models, from those that define the stability profile (refer to Section 3 – Stability Protocol Design) should
be justified and the impact of an attribute (not part of the model) changing unexpectedly should be considered as part of risk management (see Annex 2-Stability Modelling, Section 2.5-Risk Management and Model Lifecycle Considerations).
应根据模型的目的和可用的稳定性知识来选择用于建模的属性。应证明未选定用于建模的属性的合理性。选择用于建模的CQA遵循方案设计核心指南中描述的相同原则,并适应稳定性模型开发的目的。应证明从定义稳定性特征的CQA中选择模型中使用的稳定性指示CQA(参见第3节 - 稳定性方案设计)的合理性,并将属性(非模型的一部分)意外变化的影响视为风险管理的一部分(参见附录2 - 稳定性建模,第2.5节 - 风险管理和模型生命周期考虑因素)。
To establish a re-test period or shelf life, the CQAs that have been identified as those most likely to impact the product shelf life, would be selected for stability modelling, that is, those attributes that are considered to most likely approach the upper or lower bounds in the attribute specification over the storage period (ICH Q6A and 6B) and are ‘shelf life limiting’. The selected quality attributes should be justified and be the focus for developing a stability model.
为了确定复检期或有效期,将选择已确定为最有可能影响产品有效期的CQA进行稳定性建模,即认为在贮藏期间最有可能接近属性质量标准上限或下限的属性(ICH Q6A和6B),并且是“有效期限制性”属性。应证明所选质量属性的合理性,并将其作为开发稳定性模型的重点。
A2-2.2.2 Selection of Data and Parameters to Construct a Stability Model
A2-2.2.2 构建稳定性模型的数据和参数选择
The data used to build a stability model are typically based on results from the long-term primary and production stability batches or at accelerated conditions (e.g., elevated temperature and/or humidity). They may also incorporate data from earlier development studies when there is sufficient understanding of comparability between the development and production molecules.
用于建立稳定性模型的数据通常基于长期注册和生产稳定性批次或在加速条件下(例如,升高的温度和/或湿度)的结果。当对开发和生产分子之间的可比性有足够的了解时,也可以纳入早期开发研究的数据。
When limited data from the formal stability protocol are available, one may consider leveraging prior knowledge into the evaluation and model building. Prior knowledge from non-product analogous molecules may supplement the product-specific stability data. Models being developed using information from other,related products require access to sufficient prior knowledge that can be justified as transferable to the drug substance or drug product. The prior knowledge molecules that are grouped as a family or class may be justified through an evaluation of relevant characteristics for the differences between the prior knowledge molecule(s) and the drug substance or drug product. These characteristics may include structural modality, stability influencing attributes, manufacturing processes, formulation, container closure, storage conditions,analytical procedures and the available stability data, including degradation profile. Prior knowledge may be used together with primary and production batch data to generate a stability model. Any prior knowledge data from the molecule or analogous molecules should be described and structure-function differences justified, in terms of impact on a stability profile. In addition, similar analytical procedures should be used for the attributes so that the data can be appropriately transferred for inclusion in generating the stability model.
当正式稳定性方案的数据有限时,可以考虑将先验知识用于评估和模型构建。非产品类似分子的先验知识可以补充产品特定的稳定性数据。使用其他相关产品的信息开发的模型需要获得足够的先验知识,以证明其可转移到原料药或制剂中。可以通过对先验知识分子与原料药或制剂之间差异的相关特征进行评估,来证明归为一个族或类的先验知识分子的合理性。这些特征可能包括结构模式、稳定性影响属性、生产工艺、制剂、容器密封系统、贮藏条件、分析方法和可用的稳定性数据,包括降解产物谱。先验知识可以与注册和生产批次数据一起使用,以生成稳定性模型。应描述分子或类似分子的任何先验知识数据,并证明结构-功能差异对稳定性特征的影响。此外,应使用类似的分析程序来处理这些属性,以便可以适当地转移数据,并纳入在生成稳定性模型中。
When prior knowledge from analogous molecules is used in a stability model, it is important to identify and address any potential for bias that could result in over-fitting or under-fitting of the model, thereby reducing model accuracy. The management of bias in the model from the datasets used should be described.
当在稳定性模型中使用来自类似分子的先验知识时,重要的是识别和解决可能导致模型过拟合或欠拟合的潜在偏差,从而降低模型准确度。应描述所用数据集模型中的偏差管理。
The parameters (e.g., reaction rate, order of reaction) used to build a stability model should be chosen to maximise the accuracy of the inferred stability profile, while avoiding over-fitting. When prior knowledge is available, model accuracy may be assessed by using a relevant dataset that is not included in the model design for which the stability profile is known. The development of a stability model may run through several iterations while optimising the parameters to achieve a final, simplest model that provides the best prediction accuracy (least difference between the predicted value and actual experimental value).
应选择用于建立稳定性模型的参数(如反应速率、反应顺序),以最大限度地提高推断的稳定性特征的准确性,同时避免过度拟合。当先验知识可用时,可以使用未包含在已知的稳定性特征的模型设计中的相关数据集来评估模型准确度。稳定性模型的开发可能会经过多次迭代,同时优化参数,以获得最终的最简单的模型,该模型提供最佳预测精度(预测值和实际实验值之间的最小差异)。
A2-2.3 Evaluation of Data for Stability Modelling
A2-2.3 稳定性建模数据评估
The statistical approach and associated statistical parameters used should be clearly described and justified.Stability models define the trends of quality attributes that change over time, based on experimental data that may be linear or non-linear. The statistical approaches for molecule-specific stability data evaluated using linear regression and for the combining of batches for drug substance or drug product are outlined in Section 13 - Data Evaluation of the core guideline and Section 1 of this Annex. The following sections in this Annex provide additional options when using the enhanced stability models for the purpose of extrapolating the stability profile past the available data at the recommended storage conditions. The data distribution over time is typically characterised using justified statistical intervals to ensure that a defined proportion of the data lies within or that future data will lie within the interval, as appropriate for the model and statistical interval chosen.
应明确描述和证明所使用的统计方法和相关统计参数。基于可能是线性或非线性的实验数据,稳定性模型定义了随时间变化的质量属性的趋势。核心指导原则第13节 - 数据评估和本附录第1节概述了使用线性回归评估分子特定的稳定性数据的统计方法以及原料药或制剂合并批次的统计方法。本附录中的以下章节提供了使用增强稳定性模型时的其他选项,以便在推荐的贮藏条件下外推超过可用数据的稳定性特征。通常使用合理的统计区间来表征随时间变化的数据分布,以确保规定比例的数据位于该区间内或未来数据将位于该区间内,视所选择的模型和统计区间而定。
Most current enhanced stability models start with an empirical approach with the experimental stability data being compared to a mathematical or kinetic function of time. When an empirical model is used and the available stability data are being compared to the model, a demonstration of goodness of fit should be performed using appropriate statistical tools to avoid overfitting the kinetic function to the data when the model incorporates the variability and thereby reducing the accuracy of prediction.
目前大多数增强稳定性模型始于经验方法,将实验稳定性数据与时间的数学或动力学函数进行比较。当使用经验模型并将可用的稳定性数据与模型进行比较时,应使用适当的统计工具证明拟合优度,以避免当模型包含变异性时动力学函数过度拟合于数据,从而降低预测的准确性。
It is important that the accuracy of the model to infer or predict the stability profile, past the last time point of the available data, is demonstrated using appropriate statistical tools (19-27). For example, by applying the model to a known, full stability data set, for which the last timepoint result(s) has not been included, the value for that last timepoints may be predicted. The predicted value can then be compared to the known, experimentally derived value as a measure of accuracy. For quality attributes with high variability, other statistical methods of model validation should be considered because demonstration of the model’s accuracy for any single timepoint may not be sufficient.
重要的是,使用适当的统计工具(19-27)证明模型推断或预测超过可用数据的最后一个时间点的稳定性特征的准确性。例如,通过将模型应用于已知的完整稳定性数据集(其中未包括最后一个时间点的结果),可以预测该最后一个时间点的值。然后可以将预测值与已知的实验推导值进行比较,作为准确性的度量。对于变异性较高的质量属性,应考虑采用其他统计方法进行模型验证,因为仅证明任何单个时间点的模型准确性可能是不够的。
Prior knowledge data may be evaluated using Bayesian statistics as an alternative to conventional Frequentist statistics and can allow for prediction of drug substance or drug product stability data over time, past the point of available long-term condition data. The Bayesian method derives a posterior distribution for the parameters of interest by combining the likelihood distributions for the observed data with the prior knowledge. The method for derivation of the prior distribution should be justified by the applicant. The general principles outlined in this Annex for a stability model would apply to models using a Bayesian approach including verification and validation to demonstrate that the model and data used are fit for the intended purpose.
先验知识数据可以使用贝叶斯统计作为传统频率统计量的替代方案进行评估,并且可以预测超过可用长期条件数据的原料药或制剂稳定性数据随时间的变化。贝叶斯方法通过将观测数据的似然分布与先验知识相结合,推导出相关参数的后验分布。申请人应证明先验分布的推导方法的合理性。本附录中概述的稳定性模型的一般原则将适用于使用贝叶斯方法的模型,包括验证和确认,以证明所使用的模型和数据符合预期目的。
While enhanced modelling strategies are not currently associated with an upper limit for shelf life and re-test period prediction, applicants should provide risk-based and scientifically justified duration for the proposed shelf life or re-test period using enhanced approaches. When utilising enhanced stability modelling to support a shelf life or re-test period, the extent of prediction should be based on scientific understanding, risk assessment (including totality of available long-term and supportive data), prior knowledge (e.g., representative batches out to the proposed shelf life), considerations of the limits discussed in Section 13 - Data Evaluation, container closure limitations, feedback from model lifecycle considerations(e.g., emerging confirmatory data) and statistical design.
虽然增强建模策略目前与有效期和复检期预测的上限无关,但申请人应使用增强方法为拟定有效期或复检期提供基于风险且科学合理的持续时间。当使用增强稳定性模型来支持有效期或复检期时,预测范围应基于科学理解、风险评估(包括所有可用的长期数据和支持性数据)、先验知识(如达到拟定有效期的代表性批次)、对第13节 - 数据评估中讨论的限度的考虑、容器密封的局限性、模型生命周期考虑因素的反馈(如新出现的确认数据)和统计设计。
A2-2.4 Model Validation and Verification
A2-2.4 模型验证和确认
A stability model should be shown to be suitable for its intended purpose. This may be demonstrated through validation and verification procedures, for which the methodology would depend on the purpose and type of model. A comprehensive approach to model verification and validation should include discussion with experts in both analytical and statistical approaches. Model predictions may be validated by using data from earlier development studies, provided comparability has been demonstrated and the batches are considered representative of the commercial material (refer to Section 4- Selection of Batches).When a model uses accelerated condition data and the degradation kinetics obey a modified Arrhenius equation, the model may be considered as verified by fit to the modified Arrhenius equation at different storage conditions.
应证明稳定性模型适合其预期用途。这可以通过验证和确认程序来证明,具体方法取决于模型的目的和类型。模型验证和确认的综合方法应包括与专家就分析和统计方法的讨论。如果可比性已得到证明,并且批次被认为是商业批的代表,则可以使用早期开发研究的数据来验证模型预测(参见第4节 - 批次选择)。当模型使用加速条件数据且降解动力学符合修正的Arrhenius方程时,可认为该模型在不同贮藏条件下通过拟合修正的Arrhenius方程得到了验证。
Stability models are not intended to replace long-term data through the proposed re-test period or shelf life, which should be performed in addition to the model. Data should be continually obtained and evaluated, as confirmatory or ongoing verification, to assess whether the model predictions are still reliable. Models that are built using accelerated condition data may include the available long-term stability data as part of the model verification.
稳定性模型并不旨在取代在拟定复检期或有效期内的长期数据,这些数据应当与模型一同进行收集。应不断获取和评估数据,作为确认或持续验证,以评估模型预测是否仍然可靠。使用加速条件数据建立的模型可能包括可用的长期稳定性数据,作为模型验证的一部分。
A2-2.5 Risk Management and Model Lifecycle Considerations
A2-2.5 风险管理和模型生命周期考虑因素
Any stability model that infers or predicts a stability profile beyond the available drug substance or drug product data, incurs an inherent risk. A description of risk management should be provided in the regulatory submission that introduces an enhanced stability model used for setting re-test period or shelf life. The risks in using a stability model should be identified using risk management methodologies (ICH Q9) and, when applicable, appropriate mitigation strategies should be in place to reduce those risks through verification and validation activities. The resulting risk of using the stability model should be as low as possible. Use of stability models is intended for drug substances and drug products that are well understood,for which the quality attributes would be known, and their corresponding criticality and residual risks evaluated to ensure patient safety. The stability-indicating attributes are also selected, and the stability profile defined. Particular note should be taken in discussing risk presented by any use of “analogous molecules” that refers to differences between molecules that may impact their stability profile and the extent that the knowledge is transferable for use in the stability model.
任何推断或预测超出现有原料药或制剂数据的稳定性特征的稳定性模型都存在固有风险。应在提交的监管文件中提供风险管理的说明,介绍用于设定复检期或有效期的增强稳定性模型。应使用风险管理方法(ICH Q9)识别使用稳定性模型的风险,并在适用时,制定适当的缓解策略,以通过验证和确认活动降低这些风险。使用稳定性模型产生的风险应尽可能低。稳定性模型适用于已充分了解的原料药和制剂,其质量属性已知,并评估了其相应的关键性和剩余风险,以确保患者安全。也选择稳定性指示属性,并定义稳定性特征。在讨论任何使用“类似分子”所带来的风险时,应特别注意可能影响其稳定性特征的分子之间的差异,以及可转移知识用于稳定性模型的程度。
A stability model, depending on the model type and intended purpose, may require updating through drug substance and drug product lifecycle. The need to update a model should be evaluated as part of risk management. The risk assessment outcomes (from formal or informal risk management as described in ICH Q9) should also be reviewed as new data are obtained through the period that the stability model is in use.
Generally, when a model is used once to establish a re-test period or shelf life it would not be necessary to continuously update the model during lifecycle management as long as new long-term stability data are obtained that support the identified attribute trend. The post-approval and ongoing monitoring/trending of new drug substance or drug product stability data should be managed by the manufacturer’s PQS (Pharmaceutical Quality System). The PQS should be capable of detecting and managing any unexpected changes in stability trend and out of specification results with appropriate corrective action and preventive actions (CAPA) as described in ICH Q10, relevant to any stability model being used to establish re-test period or shelf life. Should an unexpected change in trend be confirmed with potential for the attribute to exceed acceptance criteria and impact the re-test period or shelf life, the model and its use should be reassessed.
根据模型类型和预期用途,稳定性模型可能需要在原料药和制剂生命周期内进行更新。更新模型的必要性应作为风险管理的一部分进行评估。在稳定性模型使用期间获得新数据时,还应审查风险评估结果(如ICH Q9所述的正式或非正式风险管理)。一般而言,当一个模型被使用一次来确定复检期或有效期时,只要获得新的长期稳定性数据支持已确定的属性趋势,就没有必要在生命周期管理期间持续更新该模型。新原料药或制剂稳定性数据的批准后和持续监测/趋势分析应由生产商的PQS(药品质量体系)管理。PQS应能够检测和管理稳定性趋势的任何非预期变化和超标结果,并采取ICH Q10中所述的与用于确定复检期或有效期的稳定性模型相关的适当纠正措施和预防措施(CAPA)。如果确认趋势发生非预期变化,且该属性有可能超出可接受标准并影响复检期或有效期,则应重新评估该模型及其使用。
Annex 3 Stability of Advanced Therapy Medicinal Products (ATMPs)
附录3 先进治疗药品(ATMP)的稳定性
A3-1 INTRODUCTION
A3-1 前言
Advanced therapy medicinal products (ATMPs) are a diverse category of innovative and complex biological products which includes somatic cell therapy, gene therapies and tissue-engineered products.ATMPs have several unique characteristics that should be reflected in the design and execution of the stability program. In some circumstances, the mechanism of action may be complex with multiple targets and potentially multiple modes of action and, as such, the critical quality attributes are not always fully understood. Owing to their complex degradation properties, accelerated stability testing conditions may not be predictive of the actual degradation profiles during storage. However, if accelerated studies can be utilised to support knowledge of the degradation profile and/or stability profile, then data and justification can be provided. The small batch size for some patient-specific ATMPs can severely limit the availability of material for stability testing. ATMPs that are designed for small patient populations may be manufactured in small batch sizes or a single batch that may even be sufficient for the entire clinical study,leading to challenges in conducting stability studies using multiple production batches. ATMPs are a class of therapeutics, which may have limited prior knowledge available to support model-based approaches to the stability assessment. In general, the shelf life for drug substance, intermediate, and/or drug product should be based on real-time stability studies.
先进治疗药品(ATMP)是一类多元化的创新和复杂生物制品,包括体细胞疗法、基因疗法和组织工程产品。ATMP有某些独特的特征,应在稳定性计划的设计和执行中得到体现。在某些情况下,其作用机制可能很复杂,有多个靶点和潜在的多种作用方式,因此关键质量属性并不总是得到充分理解。由于其复杂的降解特性,加速稳定性试验条件可能无法预测贮藏期间的实际降解产物谱。但是,如果可以利用加速研究来支持对降解产物谱和/或稳定性特征的了解,则可以提供数据和理由。一些患者特异性ATMP的小批量生产会严重限制稳定性试验材料的可用性。设计用于小患者人群的ATMP可能以小批量或单个批次生产,甚至足以满足整个临床研究,这给使用多个生产批次进行稳定性研究带来了挑战。ATMP是一类治疗药物,其可用于支持基于模型的稳定性评估方法的先验知识可能有限。一般而言,原液、中间产品和/或制剂的有效期应基于实时稳定性研究。
This annex provides recommendations for designing stability studies for ATMPs. When a topic is not included in the Annex, the reader is referred to the core guidance for stability principles that are considered generally relevant to ATMPs. The basic elements of the information detailed in Section 3 - Stability Protocol Design through to Section 14 - Labelling should serve as the basis for designing a stability program for ATMPs. For example, where an in-use period is warranted, the applicant should refer to Section 11 -In-Use Stability for general information on the principles, with the caveat that not all information in these sections may be directly relevant to ATMPs.
本附录提供了设计ATMP稳定性研究的建议。当某一议题未包含在附录中时,读者可参考通常与ATMP相关的稳定性原则的核心指南。第3节 - 稳定性方案设计至第14节 - 标签中详述信息的基本要素,应作为设计ATMP稳定性计划的基础。例如,在保证使用期间允许时限的情况下,申请人应参考第11节 - 使用中稳定性,以获取有关原则的一般信息,但注意,这些章节中并非所有信息都可能与ATMP直接相关。
A3-2 SCOPE
A3-2 范围
The recommendations in this annex apply to the assessment of stability considerations for drug substance, intermediates, and the drug products of ATMPs as appropriate depending on the product and the manufacturing process. This annex also addresses the stability considerations for starting materials (e.g., viral banks/viral seed stock). Stability considerations for reference materials used in the assessment of ATMPs are consistent with those for reference materials for other biologicals and is discussed under Section 12.1.2 - Consideration for Biological Reference Materials, of the core guidance. The document covers the generation and submission of stability data for products containing biological active substances such as autologous and allogeneic cell-based products (e.g., mesenchymal stromal cells (MSCs), Islet cells, T-cells, NK cells), xenotransplantation products (e.g., animal derived cell products), gene therapy products that are directly administered to humans (e.g., genetically modified cells, recombinant nucleic acids, viral and genetically modified bacterial vectors), oncolytic products, genome editing products and tissue engineered products.
This Annex is applicable to vectors that are administered directly as a drug product or used ex vivo to modify cells (e.g., retroviruses, adeno associated viral and other nucleic acid-based vectors) and viral banks used in the manufacture of viral and bacterial vectors.
本附录中的建议适用于评估原液、中间产品和ATMP制剂的稳定性考虑因素,具体取决于产品和生产工艺。本附录还阐述了起始物料(如病毒库/病毒种子库)的稳定性考虑因素。ATMP评估中使用的参比物质的稳定性考虑因素与其他生物参比物质的稳定性考虑因素一致,并在核心指南第12.1.2节“生物参比物质的考虑”中进行了讨论。本文件涵盖了含有生物活性物质产品的稳定性数据的生成和提交,这些产品包括基于自体和异体细胞的产品(如间充质基质细胞(MSC)、胰岛细胞、T细胞、NK细胞)、异种移植产品(如动物源性细胞产品)、直接用于人类的基因治疗产品(如转基因细胞、重组核酸、病毒和转基因细菌载体)、溶瘤产品、基因组编辑产品和组织工程产品。本附录适用于作为制剂直接给药或离体修饰细胞的载体(如逆转录病毒、腺相关病毒和其他核酸载体)以及用于制造病毒和细菌载体的病毒库。
Due to the diversity of ATMP products, the stability program should be based on process and product knowledge. The recommendations in this annex will highlight specific differences in product types, but any stability plan should consider the type of ATMP product and its manufacturing process. For example,the primary stability protocol for a vector-based gene therapy for treating larger patient populations may be different from a patient-specific cell-based therapy (i.e., personalised cellular therapies).
由于ATMP产品的多样性,稳定性计划应基于工艺和产品知识。本附录中的建议将强调产品类型的具体差异,但任何稳定性计划都应考虑ATMP产品的类型及其生产工艺。例如,用于治疗较大患者群体的基于载体的基因治疗的注册稳定性方案可能与针对特定患者的基于细胞的疗法(即个性化细胞治疗)有所不同。
A3-3 STABILITY STUDY DESIGN
A3-3 稳定性研究设计
As outlined in the core guideline, stability studies should be established based on an understanding of the product’s CQAs. ATMP stability study design should be based on process and product knowledge of the specific product type and manufacturing process. Stability testing frequency should follow the recommended testing frequency as detailed in Section 6 – Testing Frequency. When the patient specific ATMPs are stored or when the available product lot has a limited quantity, a risk-based approach to testing frequency is recommended and should be justified based on available developmental data and prior
knowledge. Stability studies for ATMPs may be performed using container closure system differing from the commercial system, when justified and supported by data showing suitability of the alternative container closure system. Shipping stability studies for ATMPs should generally follow the principles described in the core guidance. Shipping stability studies for cell based ATMPs should also include tests to evaluate the effect of physical forces exerted during shipping.
如核心指导原则所述,应基于对产品CQA的理解进行稳定性研究。ATMP稳定性研究设计应基于特定产品类型和生产工艺的工艺和产品知识。稳定性试验频率应遵循第6节 - 试验频率中详述的推荐试验频率。当贮藏了患者特异性ATMP或可用产品批次数量有限时,建议采用基于风险的检测频率方法,并应根据现有的开发数据和先验知识证明其合理性。当有数据证明替代包装容器和密封系统的适用性时,则可使用不同于商用包装容器和密封系统进行ATMP的稳定性研究。ATMP的运输稳定性研究通常应遵循核心指导原则中描述的原则。基于细胞的ATMP的运输稳定性研究还应包括评估运输过程中施加的物理力影响的试验。
The applicant is encouraged to use a risk-based approach to the design of the stability study. Where a risk-based approach is employed, the risk assessment and supporting justification should be provided.
鼓励申请人使用基于风险的方法来设计稳定性研究。如果采用基于风险的方法,应提供风险评估和支持理由。
A1-3.1 Selection of Analytical Procedures and Acceptance Criteria
A3-3.1 分析方法和可接受标准的选择
Selection of analytical procedures and acceptance criteria are detailed in the core guideline (refer to Section3.4 - Specification). Uncertainty surrounding stability CQAs due to high assay variability may be mitigated by performing orthogonal assays, where for a given CQA, orthogonal assays may provide greater confidence in the stability trends over time. Potency is a critical quality attribute for determining stability of ATMPs. However, assessing potency of some ATMPs may be challenging and complex due to incomplete knowledge of the mechanism of action of the product, absence of suitable analytical procedures
to accurately predict the product function, the inherent variability in patient-specific products, and due to the complex modes of action of the ATMP to exert a given result. Therefore, determining the change in potency during storage should be performed through a suitable assurance of the intended biological effect.
The capability of the chosen potency assay to detect subpotent or degraded product should be justified and an evaluation of the degradation profile and its impact on potency provided. When one assay is not sufficient to fully evaluate all the different product functions, multiple assays may be used to assess potency. For cell-based products, this may be evaluated through tests such as cell viability assays, immunochemistry and immunoassays for cell surface markers, and assays that evaluate function (potency). For gene therapy products, this may be evaluated through tests such as transduction, infectivity, gene expression, and/or activity of the expressed product.
分析方法和可接受标准的选择详见核心指导原则(参见第3.4节 - 质量标准)。可通过进行正交试验来缓解由于测定变异性高而导致的稳定性CQA的不确定性,其中对于给定的CQA,正交试验可为随时间变化的稳定性趋势提供更大的置信度。效价是测定ATMP稳定性的关键质量属性。然而,由于对产品作用机制的了解不全面,缺乏合适的分析方法来准确预测产品功能,患者特异性产品的固有变异性,以及ATMP产生给定结果的作用模式复杂,因此评估某些ATMP的效价可能具有挑战性和复杂性。因此,应通过对预期生物效应的适当确认来确定贮藏期间的效价变化。应证明所选效价测定法检测效价减弱或降解产物的能力,并评估降解产物谱及其对效价的影响。当仅凭一种测定法无法充分评估所有不同的产品功能时,可以使用多种测定法来评估效价。对于基于细胞的产品,可通过检测进行评估,如细胞活力测定、细胞表面标志物的免疫化学和免疫测定以及功能(效价)评估试验。对于基因治疗产品,这可以通过转导、感染性、基因表达和/或表达产品活性的测试进行评估。
The purity of the ATMP should be assessed to ensure that storage period and conditions do not lead to an increase in the levels of impurities beyond the demonstrated acceptable range. Impurities in ATMPs result from either the manufacturing process or are product related, where the latter may include for instance the following impurities: dead cells, empty viral particles, or degraded products. While process-related impurities are controlled during the manufacturing process, storage conditions and duration of storage may lead to an increase in the product-related impurities. For this reason, a quantitative enumeration of the levels
of product-related impurities in an ATMP should be assessed and the acceptable stability limits should be justified. Stability attributes related to product impurities should be based on a risk assessment at various manufacturing and storage steps (e.g., freeze-thaw step). Based on the risk, measurement of representative characteristics of the degraded/product derived material may be sufficient to assess stability, when performed in combination with other product CQAs.
应评估ATMP的纯度,以确保贮藏时限和条件不会导致杂质水平增加超过已证明的可接受范围。ATMP中的杂质来源于生产工艺或与产品相关,后者可能包括以下杂质:死细胞、空病毒颗粒或降解产物。虽然在生产工艺中控制了工艺相关杂质,但贮藏条件和贮藏时间可能导致产品相关杂质增加。因此,应评估ATMP中产品相关杂质水平的定量计数,并证明可接受的稳定性限度是合理的。与产品杂质相关的稳定性属性应基于不同生产和贮藏步骤(如冻融步骤)的风险评估。基于风险,检测降解/产品来源材料的代表性特征并结合产品其他CQA检测可能足以评估稳定性。
In addition to the general considerations for assessing stability of ATMPs, the following are examples for product-specific stability considerations that should be evaluated as a part of the stability assessment (additional product-specific parameters may also be required to assess stability):
除了评估ATMP稳定性的一般考虑因素外,以下是产品特异的稳定性考虑因素的示例,应作为稳定性评估的一部分进行评估(可能还需要其他产品特异参数来评估稳定性):
For live cell-based products that are stored frozen, it is important to measure viability of cells after they are thawed as part of the stability studies. The impact of changes in cell viability and cell concentration should be considered for subsequent processing (for intermediates) or dosing (for final product).
对于冷冻贮藏的基于活细胞的产品,评估解冻后细胞的活性是稳定性研究的重要部分。应考虑细胞活性和细胞浓度变化对后续加工(中间产品)或给药(最终产品)的影响。
For virus-based products, product CQAs such as changes to total particle number, genome copy number, infectious particle number and viral genome titre should be included in the stability studies.
对于病毒类产品,稳定性研究应包括产品CQAs,如总颗粒数、基因组拷贝数、感染性颗粒数和病毒基因组滴度的变化。
For viral therapy vectors that are used to further modify cells ex vivo, vector integrity, potency and strength are stability-indicating CQAs that should normally be included in stability studies.
对于用于进一步体外修饰细胞的病毒载体,载体完整性、效价和强度是稳定性指示性CQAs,通常应包括在稳定性研究中。
For bacteria-based products, the viability, bacteria count, plasmid copy number (if applicable)should be considered.
对于细菌类产品,应考虑活性、细菌计数、质粒拷贝数(如适用)。
For DNA or RNA based products, stability determination may also include an assessment of
structural integrity and quantity in addition to other purity assessments.
对于基于DNA或RNA的产品,除了其他纯度评估外,稳定性测定还可能包括结构完
整性和数量评估。
For Tissue Engineered products physicochemical and functional critical quality attributes should be evaluated as a part of the stability studies. The product’s structural stability may be evaluated through tests such as measurement of size and shape, and assessment of structural integrity. In the case of products formulated with carrier or support materials, the stability of the complex formed with the drug substance should be studied.
对于组织工程产品,应将理化和功能关键质量属性作为稳定性研究的一部分进行评估。可通过尺寸和形状测量、结构完整性评估来评估产品的结构稳定性。对于使用载体或支撑材料配制的产品,应研究与原液形成的复合物的稳定性。
Acceptance criteria should be justified considering the data from material used in preclinical and clinical studies. For substances that cannot be properly characterised or products for which an exact analysis of the stability-indicating CQAs cannot be determined through routine analytical procedures, the applicant should propose and justify alternative testing procedures. The attributes tested for batch release may not be entirely suitable for stability determination for some ATMPs (e.g., definition of mature and immature dendritic cells
based on the surface expression of markers such as CD80, CD86, CD83, and MHC II; percent (%) transduced products in case of an ex vivo modified cellular product; virus phenotype and genetic identity of virus). Acceptance criteria for acceptable impurities should be derived from the analytical profiles of batches of the drug substance and drug product used in the preclinical and clinical studies, and batches that did not adversely impact safety or potency should be used to set acceptance limits for impurities. When justified, shelf life specifications may differ from the release specification.
考虑到临床前和临床研究中使用的材料数据,可接受标准应合理明确。对于无法正确表征的物质或无法通过常规分析方法确定稳定性指示性CQAs精确分析的产品,申请人应提出并证明替代检测方法的合理性。批放行检验的属性可能不完全适用于某些ATMP的稳定性测定(例如,基于CD80、CD86、CD83和MHC II等标志物的表面表达来定义成熟和未成熟的树突状细胞;离体修饰细胞产品中转导产品的百分比(%);病毒表型和病毒的遗传特性)。杂质的可接受标准应来源于临床前和临床研究中使用的原液和制剂批分析图谱,应使用对安全性或效价没有不利影响的批次来设定杂质的可接受标准。在合理的情况下,有效期质量标准原则上可能与放行质量标准不同。
A1-3.2 Selection of Study Conditions
A3-3.2 研究条件的选择
Recommendations around the selection of study conditions are outlined in the core guideline (refer to Section 3 – Stability Protocol Design through Section 7 – Storage Conditions). It is expected that the stability studies for ATMPs include real-time storage and in-use period conditions. Generally, accelerated or stress testing may not provide direct information to support shelf life, however testing under accelerated and stressed conditions are recommended for ATMPs. Accelerated studies may be utilised to gain knowledge of the stability profile. Testing under these conditions help in the determination of the extent of
temperature deviations that can be tolerated while the harsher stress conditions may provide information on the degradation profile of the product. Accelerated or forced degradation studies can also be useful to demonstrate the stability-indicating nature of assays and their corresponding levels of sensitivity. The accelerated conditions defined in Section 7 - Storage Conditions of the core guideline may not be directly applicable for ATMPs, and the accelerated and stressed conditions should be carefully selected based on a risk assessment and worst-case conditions relevant to ATMP’s handling and storage.
核心指导原则中概述了关于研究条件的选择的建议(参见第3节 - 稳定性方案设计至第7节 -贮藏条件)。预计ATMP的稳定性研究包括实时贮藏和使用条件。一般而言,加速或强制降解试验可能无法提供支持有效期的直接信息,但建议在加速和强制降解条件下对ATMP进行检测。加速研究可用于获取稳定性特征的知识。在这些条件下进行试验有助于确定产品可容忍的温度偏差范围,而更严苛的强制降解条件则可以提供关于产品降解产物谱的信息。加速或强制降解研究也可用于证明稳定性指示分析的性质及其相应的灵敏度水平。核心指导原则第7节“贮藏条件”中定义的加速条件可能不直接适用于ATMP,应根据风险评估和与ATMP处理及贮藏相关的最坏情况谨慎选择加速和强制降解条件。
A1-3.3 Selection of Batches
A3-3.3 批次选择
Recommendations around the selection of batches are outlined in the core guideline. Generally, stability data from 3 primary batches are recommended to support the proposed shelf life of ATMPs, however on the basis of risk evaluation alternative number of stability batches may be justified. The risk to an accurate determination of predicted shelf life of an ATMP will depend on various factors including the assay limitations and variabilities in and the quality of starting materials. The shelf life of ATMPs should generally be justified based on long-term stability data through the proposed shelf life. In some instances,
a stability profile based on prior knowledge from analogous products (refer to Annex 2-Stability Modelling) may provide additional supporting stability data. As described in the core guideline Section 4.1 -Considerations for Selection of Primary Stability Batches, the manufacturing scale of the primary stability batches for ATMPs may differ from that of the production batch, unless the scale change represents a significant risk to stability. Primary stability batches may be clinical batches not at production scale, provided appropriate comparability has been demonstrated to production batches. When primary batches are not production scale batches, a post-approval commitment may be required to confirm the stability.
核心指导原则中概述了关于批次选择的建议。一般来说,建议使用3个注册稳定性批次的稳定性数据来支持ATMP的建议有效期,但是根据风险评估,可以证明稳定性批次的替代数量是合理的。准确确定ATMP的预测有效期的风险将取决于各种因素,包括方法学限度及起始物料质量的变异性。通常应根据建议有效期内的长期稳定性数据证明ATMP保质期的合理性。在某些情况下,基于类似产品的先验知识的稳定性特征(见附录2-稳定性建模)可提供额外的支持性稳定性数据。如核心指导原则第4.1节“注册稳定性批次选择的考虑因素”所述,ATMP注册稳定性批次的生产规模可能与生产批次不同,除非规模变化对稳定性构成重大风险。注册稳定性批次可以是非生产规模的临床批次,前提是已证明与生产批次具有适当的可比性。当注册稳定性批次不是生产规模批次时,可能需要批准后承诺来确认稳定性。
Stability of patient-specific cellular ATMPs should be obtained from patient derived materials. However, this may not always be feasible due to limited availability (e.g., autologous CAR-T cells), and when justified, stability data from representative healthy donor derived materials along with stability data from patient-derived material may be acceptable. When patient derived material is only available in limited quantities to perform the recommended stability studies as per the stability protocol, the principles of bracketing (refer to Annex 1 - Reduced Stability Protocol Design) may be applied to ATMPs. Stability-indicating CQAs that are assessed to determine stability will depend on the drug product and should be justified.
患者特异性细胞ATMP的稳定性应从患者来源的材料中获得。然而,由于可及性有限(比如自体CAR-T细胞),该方法可能并不总是可行的,且在合理的情况下,可接受来自健康供体来源的代表性材料的稳定性数据以及基于患者来源的材料的稳定性数据。根据稳定性方案进行推荐的稳定性研究时当患者来源材料有限,括号法原则(见附录1-简化稳定性方案设计)可适用于ATMP。用于确定稳定性的稳定性指示性CQA将取决于制剂,并应证明其合理性。
Stability of cryopreserved cells used as cell substrates for the manufacture of ATMPs may apply the principle of modelling based on prior knowledge (e.g., cell type, formulation, container, cell density) to set initial shelf life beyond long-term data at submission.
ATMPs may differ in the standard manufacturing process flow without having distinct bulk drug substance batches and manufactured in one uninterrupted stream with no distinct drug substance storage step. Under such circumstances, there would not be a need to evaluate the storage period of a drug substance. Examples of this type of manufacturing include a number of cell-based products that are continuously cultured,purified, formulated and stored (or administered fresh) as the final ready-to-use drug product.
冻存细胞(用于ATMP生产的细胞基质)的稳定性可以采用基于先验知识(如:细胞类型、剂型、容器、细胞密度)的建模原理,以在提交时设定超过长期数据的初始有效期。
ATMP与标准生产工艺流程有所不同,它们可能没有具体的原液批次,而是在一个不间断的流程中生产,并且没有明确的原液贮藏步骤。在这种情况下,不需要评估原液的贮藏期。此类生产的例子包括许多基于细胞的产品,这些产品作为最终即用型制剂会经过连续培养、纯化、配制和贮藏(或新鲜给药)。
For ATMPs that have a distinct drug substance stage, the date of manufacturing of the drug substance and the date of manufacturing of the drug product may be two separate dates and the duration of storage of the drug substance prior to it being processed into the final drug product may influence the storage period of the drug product. If this is the case, a risk assessment should be performed to determine if the stability assessments should also take into consideration the cumulative storage period of the drug substance and drug product.
When the manufacturing process includes a short hold time, a risk assessment to determine the need and extent of hold time stability studies should be assessed and justified. Some ATMP manufacturing process may include a freezing step (e.g., short term storage of cells prior to further processing). In such instances,the stability of the stored intermediate should be evaluated upon thaw.
对于具有不同原液阶段的ATMP,原液的生产日期和制剂的生产日期可能是两个不同的日期,原液在加工成最终制剂之前的保持时限可能会影响制剂的贮藏期。如果是这种情况,应进行风险评估以确定稳定性评估是否还应将原液和制剂的累积贮藏期纳入考虑。
当生产工艺包含较短的保持时限时,应评估并证明风险评估以确定保持时间稳定性研究的必要性和范围。一些ATMP生产工艺可能包括冷冻步骤(比如在进一步处理之前短期贮藏细胞)。在这种情况下,应在解冻后评估贮藏中间产品的稳定性。
A3-4 STARTING MATERIALS AND STABILITY
A3-4 起始物料和稳定性
The protocol should take into consideration that stability of ATMPs may be affected by the quality of starting materials and viral vectors, and stability assessment should consider the impact of starting materials for cell therapy products (e.g., allogenic, autologous cells), transport, storage steps in the manufacturing process, and their short or long-term storage conditions (e.g., short-term cell storage versus long-term cryopreservation). The stability of cellular starting materials (e.g., donor cells) should be assessed during their storage and shipping. In general, assessment of stability of cellular starting materials during their
storage and shipping should follow the recommendations detailed in this guideline for cell based ATMPs and should follow a risk based approach to determining their stability. Stability of starting materials used to manufacture gene therapy vectors (e.g., plasmids, virus banks used to make vectors) should also be controlled.
稳定性方案应考虑到ATMP的稳定性可能受到起始物料和病毒载体质量的影响,稳定性评估应考虑细胞治疗产品的起始物料(如异体细胞、自体细胞)、运输、生产工艺中的贮藏步骤及其短期或长期贮藏条件(如短期细胞贮藏与长期冻存)的影响。应评估细胞起始物料(如供体细胞)在贮藏和运输期间的稳定性。一般来说,细胞起始物料在贮藏和运输过程中的稳定性评估应遵循本指导原则中关于基于细胞的ATMP的详细建议,并应遵循基于风险的方法来确定其稳定性。还应控制用于生产基因治疗载体的起始物料(如质粒、用于制造载体的病毒库)的稳定性。
Viral vectors that are used to modify cells ex vivo to make ATMPs (e.g., retrovirus, lentivirus) are normally manufactured in bulk, purified and adjusted for a desired concentration and stored frozen until use. Stored viral vectors should be assessed for stability related CQAs, such as vector integrity, strength (e.g., infectious titre, transducing titre, genomic titre and viral particle count), product related impurity profile, ratio of empty to full particles (if applicable), activity (e.g., gene expression), and sterility (or container closure integrity testing). When the viral vectors are stored at varying concentrations, stability of the viral vectors at each individual concentration should be assessed, unless bracketing is justified (refer to Annex 1- Reduced Stability Protocol Design).
用于ATMP生产的进一步体外修饰细胞的病毒载体(如逆转录病毒、慢病毒)通常以批量生产,经纯化并调节至所需浓度后,被冷冻贮藏直至启用。应评估贮藏的病毒载体的稳定性相关CQA,包括载体完整性、强度(如:感染性滴度、转导滴度、基因组滴度和病毒颗粒计数)、产品相关杂质谱、空粒子与完整粒子的比率(如适用)、活性(如基因表达)和无菌性(或容器密封完整性检测)。当病毒载体在不同浓度下贮藏时,除非能够证明使用括号法是合理的,应评估每个浓度下病毒载体的稳定性(见附录1-简化稳定性方案设计)。
A3-4.1 Cell and Viral Banks
A3-4.1 细胞库和病毒库
The stability of cell banks under defined storage conditions should be generated to verify that the thawed cells have survived the preservation process and retain their CQAs, consistent with the recommendations outlined in ICH Q5D. A stability protocol for monitoring of banked cells should be provided in the submission. Stability-indicating CQAs that are assessed to determine stability should be justified.
应根据ICH Q5D中概述的建议,评估细胞库在规定贮藏条件下的稳定性,以验证解冻后的细胞在贮藏过程中存活并保留了其CQA。应在申报资料中提供用于监测入库细胞的稳定性方案。应证明为确定稳定性而评估的稳定性指示性CQA的合理性。
Stability of viral banks that are either used in the production of viral drug products for direct administration or for viral vectors used in the production of in vitro modification of cells should be evaluated for stability.
应评估病毒库的稳定性,这些病毒库可用于生产直接给药的病毒制剂或用于生产体外修饰细胞的病毒载体。
The quality of the viral bank should be well established and would typically include an evaluation of its stability-indicating CQAs. When establishing the stability period of viral banks, virus stability may be demonstrated in some cases through assessing the quality attributes of the drug substance, manufactured from the stored material at the end of the viral bank’s shelf life. The stability of the established master viral banks (also referred to as viral seed stock in some regions) and working viral banks should be evaluated periodically per a stability protocol. The stability protocol should describe and justify the test parameters
and stability acceptance criteria which should be based on its intended use. Potency may also be a stability-indicating CQA, depending on the intended use of the viral bank (e.g., when used to manufacture a viral drug product). Depending on the intended use of the viral bank (e.g., when used to manufacture a viral drug product), infectious titre may also be a stability-indicating CQA and should be included as a part of viral bank’s stability assessments.
应充分确定病毒库的质量,其中通常包括对其稳定性指示性CQA的评估。在建立病毒库的稳定期时,在某些情况下,可以通过评估由贮藏材料在病毒库有效期结束时生产的原液的质量属性,来证明病毒的稳定性。应根据稳定性方案定期评估已建立的主病毒库(在某些地区也称为病毒种子库)和工作病毒库的稳定性。稳定性方案应描述并证明基于其预期用途的试验参数和稳定性可接受标准。根据病毒库的预期用途(比如当用于生产病毒制剂时),效价也可能是稳定性指示性CQA。根据病毒库的预期用途(比如当用于生产病毒制剂时),感染性滴度也可能是稳定性指示CQA,应该被纳为病毒库稳定性评估的一部分。
A3-5 ESTABLISHMENT OF SHELF LIFE
A3-5 有效期的建立
The shelf life of ATMPs may not be accurately predicted from accelerated stability studies, as their behaviour can vary considerably based on the temperature and related changes in the storage medium.When the accelerated stability studies only provide a limited information, due to differences in degradation profile, stability studies designed to support product stability should be performed in real time, under the intended storage conditions. When sufficient real-time stability data from the production lot is not available for ATMPs, stability data from developmental batches and prior knowledge from similar products may be used as supporting data to justify setting initial stability period, with a concurrent testing strategy built into the stability testing protocol. The use of prior knowledge to support shelf life determination of an ATMP should be discussed with regulatory authorities as appropriate.
根据加速稳定性研究,可能无法准确预测ATMP的有效期,因为其特性可能会根据贮藏介质的温度和相关变化而发生很大变化。当加速稳定性研究提供的信息有限时,由于降解产物谱上的差异,应在预期贮藏条件下进行旨在支持产品稳定性的实时稳定性研究。当生产批次中没有足够的实时稳定性数据可用于ATMP时,开发批次的稳定性数据和类似产品的先验知识可用作支持性数据,以证明设定初始稳定期的合理性,并在稳定性试验方案中建立同步试验策略。使用先验知识来支持ATMP有效期的确定,并应酌情与监管机构讨论。
A minimum of 6 months stability data should be included at the time of submission. The shelf life may be extended beyond the initial 6-month period when additional stability data becomes available. For drug products with storage periods of less than 6 months, the minimum amount of stability data in the initial regulatory submission should cover the intended shelf life.
申报时应包括至少6个月的稳定性数据。当获得额外的稳定性数据时,有效期可延长至超过最初的6个月。对于有效期少于6个月的制剂,首次注册申报的最低限度稳定性数据应涵盖预期的有效期。
ATMPs that have a storage period at the drug substance stage and at the drug product stage should be assessed for stability under the stability protocol as detailed in Section 3 - Stability Protocol Design of the core guidance. When intermediates used in the manufacture of cellular products and viral vectors are stored, they should also be assessed for their stability under a pre-specified stability program and a shelf life established based on real-time stability information.
对于在原液阶段和制剂阶段有贮藏期的ATMP,应根据核心指导原则第3节“稳定性方案设计”中详述的稳定性方案对其进行稳定性评估。当贮藏用于细胞产品和病毒载体生产的中间产品时,还应在预先规定的稳定性计划下评估其稳定性,并根据实时稳定性信息确定有效期。

来源:国家药监局审评中心
关键词: 稳定性试验