
嘉峪检测网 2025-06-12 08:34
导读:该指导原则重点介绍了如何利用亚硝胺类杂质的结构特点预测其致癌性,并根据致癌潜力类型确定该杂质可接受摄入量限度的方法。
摘要
亚硝胺类药物相关杂质(NDSRI)与原料药(API)具有结构相似性(在化学结构中具有API或API片段),是每种原料药特有的杂质。NDSRI通常是由药物制剂中含有仲胺或叔胺的API(或API片段)在接触亚硝化剂(如制剂辅料中的残留亚硝酸盐)时形成。NDSRI通常缺乏致癌性和致突变性试验数据,因此无法根据该数据确定可接受摄入量限度。美国美国食品药品监督管理局(FDA)于2023年8月发布了“亚硝胺类药物相关杂质推荐可接受摄入量限度行业指导原则”,该指导原则重点介绍了如何利用亚硝胺类杂质的结构特点预测其致癌性,并根据致癌潜力类型确定该杂质可接受摄入量限度的方法。详细介绍FDA该指导原则的前言、背景,根据该指导原则提出了根据预测的致癌性分类确定可接受摄入量限度内容和对NDSRI推荐的可接受摄入量限度的实施的具体建议,期望能为我国亚硝胺类杂质可接受摄入量限度的研究和监管提供一定的参考。
【关键词】美国食品药品监督管理局;亚硝胺类药物相关杂质;致癌性,可接受摄入量限度;监管
美国食品药品监督管理局(FDA)于2023年8月7日发布了指导原则“亚硝胺类药物相关杂质(nitrosamine drug substance related impurities,NDSRI)推荐可接受摄入量限度行业指导原则”[1],该指导原则由FDA药物评价与研究中心(CDER)组织发布。FDA在实施本指导原则前并未事先征求公众意见,因为FDA认为,由于这类亚硝胺杂质在多种药物制剂中发现,也可能存在于原料药(API)中,对于药品申请人和生产企业而言,及时提供该议题的补充信息非常重要,因此让公众提前参与评论并不可行或不合适。自FDA《人用药品亚硝胺类杂质控制行业指导原则》发布以来,FDA已经收到了关于NDSRI的报告。NDSRI与原料药具有结构相似性(在化学结构中具有API或API片段),是每种API特有的杂质。根据原料药的化学结构,在大量药物制剂中存在形成NDSRI的风险。因此该指导原则在FDA发布之日起执行,但仍会根据FDA的优良指导原则规范接受各方意见。
我国于2020年5月8日发布了《化学药物中亚硝胺类杂质研究技术指导原则(试行)》[2],该指导原则主要涉及工艺产生、降解途径和污染引入等途径产生的亚硝胺杂质,未涉及NDSRI。国际人用药品注册技术协调会(ICH)于2024年6月成立了M7亚硝胺亚组,计划在2030年3月前完成亚硝胺相关杂质的国际协调。本文将详细介绍FDA“亚硝胺类药物相关杂质推荐可接受摄入量限度行业指导原则”主要内容,以期为我国亚硝胺类药物相关杂质可接受摄入量限度制定及药物开发评价给予一定的启发和帮助。
1.该指导原则的前言
该指导原则为药品生产企业和申请人提供了一种用于预测药品中NDSRI可能存在的致突变性和致癌性潜力的原则,并对NDSRI的可接受摄入量(acceptable intake,AI)限度给出了建议。NDSRI通常是由制剂中含有仲胺或叔胺的API(或API片段)在接触亚硝化剂(如制剂辅料中的残留亚硝酸盐)时形成。NDSRI通常缺乏致癌性和致突变性试验数据,因此无法根据该数据确定AI限度。本指导原则提供了一种利用NDSRI的结构特征预测其致癌潜力类型并根据不同的类型推荐相应AI限度的方法。
由于对药品中NDSRI的存在和可接受水平具有不确定性,因此对该类药品的监管具有一定的挑战性,可能导致一些申请人进行不必要的研究或在某些情况下停售药品。此外,由于在许多药品中发现亚硝胺杂质,可能导致药物因供应中断而出现药品短缺。
FDA建议对NDSRI进行基于风险的安全性评估。生产企业和申请人可结合该指导原则以及FDA于2021年2月发布的《人用药中亚硝胺杂质控制行业指导原则》[3]的建议确定其制剂和原料药中NDSRI的AI限度。FDA认为,FDA推荐的AI限度以及生产企业和申请人得出的AI限度可能会随着亚硝胺学科发展以及研究进展而变化。随着业界对亚硝胺杂质的致突变性和致癌性风险的认识增加,FDA鼓励业界分享此类研究信息以帮助扩展现有的知识库。
该指导原则适用范围包括已批准或待批准的新药注册申请(NDA)或仿制药注册申请(ANDA)所涉及的处方药和非处方药,以及未通过药品申请而销售的产品,如《联邦食品,药品和化妆品法》第505G节的非处方药,或符合现行良好生产规范(cGMP)的其他药品。本指导原则也适用于临床开发中的处方药和非处方药,还适用于含有化学合成片段的生物制品或含有药物成分的生物主导组合产品。本指导原则不适用于晚期抗肿瘤药中的NDSRI[此类药物可参考ICH S9、ICH Q3A(R2)、ICH Q3B(R2)]。
2.该指导原则的背景
自2018年FDA获知缬沙坦[4]中存在一种N-亚硝基甲胺(NDMA)杂质以来,FDA一直致力于解决药品中亚硝胺杂质的评价和监管问题。药品中最初报告的亚硝胺杂质是小分子亚硝胺,如NDMA,该化合物是由胺(仲胺、叔胺或季胺)和亚硝酸盐通过亚硝化反应形成,通常是由于在原料药合成时加入了亚硝酸、亚硝酸盐和酰胺溶剂。2020年,FDA发布了《亚硝胺指导原则》,建议API和制剂生产企业采取相应的措施对杂质进行检测,防止药品中的亚硝胺杂质含量超标或尽量避免亚硝胺杂质的产生。《亚硝胺指导原则》建议生产企业和申请人应采取三步法以减少药品中亚硝胺杂质:(1)对其API和制剂中的亚硝胺进行风险评估;(2)发现风险后进行确证性检测;(3)对已经批准或正在审评的NDA和ANDA品种,为避免或减少原料药和制剂中亚硝胺杂质的产生而实施的变更应当向FDA报告。根据《亚硝胺指导原则》,API和制剂生产企业应采取适当措施以防止亚硝胺杂质超标。由于亚硝胺类化合物对多种动物种属具有潜在的遗传毒性,有些被归类为潜在的致癌物,因此在《ICH M7(R2)评估和控制药物中的DNA活性(致突变)杂质以限制潜在的致癌风险》(2023年7月)中,将它们归入关注队列(cohort of concern)。ICH M7(R2)指导原则提供了控制已知或潜在致突变致癌物的建议,如硝基化合物,保证人体在一定的暴露水平下致癌性风险基本可忽略不计[5]。
在AI限度水平下,保守假设人一生(70年)中每日都暴露于该杂质,每10万人中会增加1例患癌症风险。换句话说,AI限度是指每天接触一种化合物如NDMA,在接触70年后,患癌症的风险约为1∶100 000。与人类一生中罹患任何类型癌症的总发病率(高于1/3)相比,这一风险在理论上只是小幅增加。如果已明确亚硝胺杂质具有潜在风险,并且已通过适当的检测方法确认了亚硝胺水平,则应制定合理的控制策略,以确保亚硝胺杂质保持在或低于AI限度。根据ICH M7(R2)的建议,AI限度通常基于数据库和文献搜索到的致癌性和细菌致突变数据来确定。
随着FDA对NDRSI的认识不断深入,该指导原则建议对NDSRI进行基于风险的安全性评估。该指导原则中提出的预测致癌性潜力分类方法(CPCA)是对ICH M7(R2)中提供方法的补充。它基于ICH M7(R2)的原则,建议使用结构-活性关系(SAR)对杂质的致突变性和致癌性风险进行评估和分类,以限制潜在的致癌性风险。由于对N-硝基化合物的SAR的科学研究已取得一定进展,因此,FDA建议使用这种预测方法来评估NDSRI的致癌性,并更准确地预测其致突变潜力,对于不同致癌性分类的NDSRI将推荐不同的AI限度。
确定NDSRI的AI限度通常比确定小分子亚硝胺的AI限度更具挑战性,因为对每种API来说NDSRI都是独特的,所以NDSRI的安全性数据(如啮齿类动物致癌性数据)有限。FDA已经对有限数量的NDSRI发布了推荐的AI限度,但与更常见的亚硝胺不同,目前大多数NDSRI尚未确定推荐的AI限度。
AI限度的确定需要基于安全性评估,并且AI限度需维持或低于FDA已确定的不会对患者造成安全性担忧的杂质水平。一种化合物特定的AI可根据已发表的科学文献中啮齿类动物致癌性数据计算,例如TD50值(该剂量下导致肿瘤发生率达到50%,相当于癌症风险概率1/2)。致癌性数据可从致癌性数据库(CPDB)或Lhasa致癌性数据库(LCDB)获取。约有140个亚硝胺类化合物(大多数为小分子亚硝胺)的TD50值最早在CPDB中报告,并已被纳入LCDB。这些化合物的致癌效力值差异较大(范围超过4个数量级),并且有一些亚硝胺为非致癌物质[6]。然而,很多研究数据并不可靠,所以不能仅依靠这些研究来确定AI限度。
当NDSRI的致突变性没有得到充分表征时,FDA和申请人支持使用定量(Q)SAR方法确定一种经可靠方法检测过的替代化合物(其结构和反应性与NDSRI相似)以此预估致癌性,根据此方法也可以得到具有科学性的AI限度。在这种情况下,替代化合物在与NDSRI相同的化学环境中应含有N-硝基警示结构,并且具有可靠的致癌性数据[7-9]。替代品的选择依据非常重要,因为从替代化合物中获得的数据将用于对缺少相关数据的化合物进行定量或定性的评估(通常称为交叉参照评估,read-across analysis)。
在选择合适的参比化合物进行交叉参照评估时,NDSRI的N-亚硝基基团的周围结构环境是一个重要考虑因素,这些结构环境可能包括取代度、空间体积、电子的影响、代谢活化潜力、产生的代谢物的稳定性/反应性和总相对分子质量。
3.根据预测的致癌性分类确定AI限度
3.1 预测CPCA简介
该指导原则推荐了一种可根据NDSRI的激活和失活结构特征预测NDSRI的致癌性分类从而确定AI限度的方法。激活或失活的结构特征分别指增加或降低NDSRI致癌性的分子亚结构。该方法参考了近年来文献发表的亚硝胺化合物SAR概念[7-9],认为α-羟基化代谢激活机制[10]是导致许多亚硝胺化合物致突变和强致癌性的原因,因此可根据NDSRI的结构特征来预测其致突变和致癌潜力,认为含有直接促进或抑制代谢激活的结构特征,或可通过其他生物途径加快亚硝胺清除的结构特征将对NDSRI致癌性产生相应的影响。
该指导原则中提出的预测致癌性分类方法适用于N-亚硝胺基团两侧结合一个碳原子的NDSRI,这些碳原子不直接通过双键与杂原子连接(例如N-亚硝基酰胺、N-亚硝基脲、N-亚硝基胍和其他类似分子结构,都不包括在该指导原则内)。此外,该方法不适用于N-亚硝胺基团在芳香环结构内的NDSRI(例如亚硝基吲哚)。
该指导原则中提出的预测致癌性方法是基于CPDB和LCDB中公布的小分子亚硝胺数据信息,同时参考了在近期文献中发表的亚硝胺化学和生物机制考虑因素[6,11]。在开发该模型时主要根据以下几个方面确定激活和失活结构特征权重,如已发表文献中报告的SAR趋势,在CPDB和LCDB中观察到的SAR趋势,以及对化合物的化学和生物机制途径的一般科学性解读。NDSRI可应用基于结构特征的方法来预测NDSRI的相对致癌性,相比于采用亚硝胺替代分子确定AI,这种方法考虑了分子中多种特征的影响,并利用了已发表的科学文献中更多的致癌性数据。FDA预计对这些结构特征的分析以及对这些特征的权重分配,可能会随着科学的进步和积累数据的增多而发生改变。
FDA认为,该方法不适用于FDA在某些情况下推荐的AI限度(例如,基于特定化合物评估或替代化合物交叉参照评估)。通常,FDA会通过直接与申请人或生产企业沟通或通过FDA指导原则(亚硝胺指导原则)来讨论推荐的AI限度。当FDA根据特定化合物评估或替代化合物交叉参照评估发布推荐的AI限度时,生产企业和申请人应当采用FDA推荐的AI限度,而非该指导原则中的预测致癌性分类方法来确定推荐的AI限度。
该指导原则中描述的预测致癌性分类方法是一种保守的方法,代表了当前最具科学性的方法,并有望随着新数据的获得而进一步完善和扩展。此外,同所有指导性文件一样,在满足适用的法规要求下,生产企业和申请人也可以使用其他替代方法。
3.2 预测CPCA的具体应用
FDA建议根据NDSRI预测CPCA制定以下AI限度。表1展示了预测的致癌性分类和相应建议的AI限度。
▲表1- NDSRI 的 5 种预测致癌性分类和相关推荐 AI 限度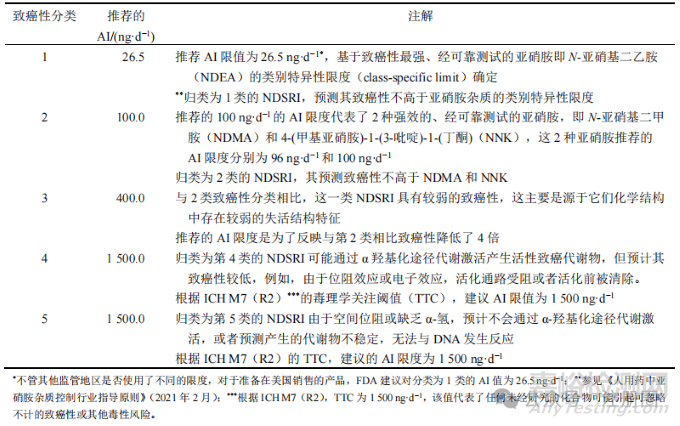
3.3 预测CPCA和推荐相应的AI限度
图1所示的流程图根据NDSRI中激活和失活结构特征展示了将NDSRI归类为不同致癌性分类的过程,并推荐了相应的AI限度。该指导原则附录A[1]提供了2个案例展示如何对致癌效力评分并确定相应的致癌性分类。流程图中NDSRI中α-和β-碳是相对于N-亚硝基定义的,如图2所示。
如上所述,该指导原则中描述的预测致癌性分类和推荐的AI限度方法适用于N-亚硝基两侧均含有碳原子的NDSRI,其中碳原子不直接与杂原子双键连接(例如N-亚硝酰胺、N-亚硝基脲、N-亚硝基胍和其他相关结构除外)。此外,致癌性分类方法不适用于N-亚硝基位于芳香环内的NDSRI(例如亚硝化吲哚)。图1描述了如何预测NDSRI的致癌性分类,并推荐了相应的AI限度。对于含有2个N-亚硝基的NDSRI,应选择预测致癌性最高的基团来确定整个分子的AI限度。
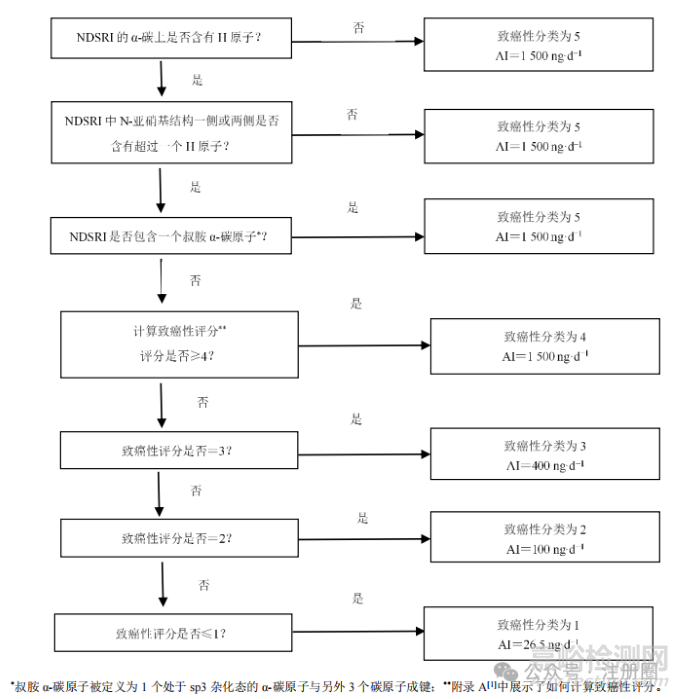
▲图1-预测 NDSRI 致癌性分类并确定相关推荐 AI 限度的流程图
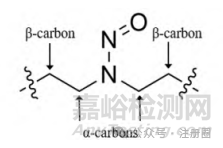
▲图 2-NDSRI 中 α-和 β-碳的结构表征
3.4 对药品含有多种亚硝胺的生产厂家和申请人的建议
FDA指出,NDSRI推荐的AI限度是针对药物中单一的NDSRI。如果药物中检测到多个亚硝胺杂质(多个NDSRI和/或小分子亚硝胺),并且根据每日最大剂量推算,亚硝胺的总水平超过了药物中致癌性最强的亚硝胺的推荐AI限度,生产企业或申请人应与FDA联系。如果单个亚硝胺的推荐AI限度差异很大,那么根据致癌性最强的单个亚硝胺限度来确定总亚硝胺限度可能不切实际,在这种情况下,也鼓励申请人与FDA沟通,讨论亚硝胺杂质限度问题。
3.5 针对某些API中存在生成NDSRI风险的推荐AI限度
经确认,一些含有仲胺或叔胺的API可能存在生成NDSRI风险。假设在特定的制剂处方和生产工艺条件下,例如药物辅料中含有残留的亚硝酸盐,这些API可能会生成NDSRI。FDA将基于对化学结构的分析,得出当API中含有亚硝化形式的仲胺和二甲基叔胺基团时,可以对API中形成的NDSRI采用预测致癌性分类方法推荐AI限度的结论[12]。
4.对NDSRI推荐的AI限度的实施(包括采用CPCA方法)
4.1 NDSRI的推荐实施时间线
FDA期望生产企业和申请人参照《亚硝胺指导原则》中第III和V节中描述的三步缓解策略来确定是否存在亚硝胺杂质包括NDSRI,但《亚硝胺指导原则》中推荐的时间线不适用于与NDSRI相关的风险评估、确证性检测和提交变更。对于NDSRI,FDA根据药品的监管状态为生产企业和申请人推荐了不同的实施时间线。
4.1.1 对于已批准或上市药品的推荐时间线FDA已了解到,一些生产企业和申请人已经考虑了在《亚硝胺指导原则》中最初确定的亚硝胺杂质,但并未对NDSRI进行风险评估。FDA建议,如果之前并未对NDSRI进行风险评估,作为整体风险管理的一部分,生产企业和申请人应在本指导原则发布后3个月内重新评估风险,建议在2023年11月1日之前完成评估。一旦确定了NDSRI的风险,应尽快采用灵敏且经过适当验证的方法进行确证性检测,对于具有高风险的药品应立即进行确证性检测。如果在药物中检测到的NDSRI水平超过了推荐的AI限度,FDA建议生产企业和申请人制定控制策略和/或设计方法,控制NDSRI在可接受的水平内。FDA建议在2025年8月1日前完成药品NDSRI确证性检测,同时在药品申请时提交必要变更。至2025年8月1日,生产企业和申请人应确保其药品中NDSRI符合FDA推荐的AI限度要求。
FDA认为实施时间线包括调查NDSRI杂质产生的根本原因,确定适当的有效变更(例如,变更生产工艺、成分供应商、制剂处方等)[13],并证明变更将尽可能减少NDSRI的产生而不会对药品质量产生不利影响。FDA可根据现有信息要求申请人进行加快的风险评估、确证性检测或采取其他监管措施。
如果确定药品中存在NDSRI风险,则应采用灵敏且经过适当验证的方法对各批次进行确证性检测。如果检测到亚硝胺杂质,生产企业应研究亚硝胺杂质产生的根本原因,并根据《亚硝胺指导原则》中的建议,对生产工艺或制剂处方实施变更,以彻底避免或减少亚硝胺杂质的产生。生产企业必须按照适当的要求实施变更。此外,申请人必须通过补充或修正申请的方式提交拟订的制剂变更。一般而言,FDA认为已上市药品的制剂处方变更为重大变更,此类变更需要提交事先批准补充申请(PAS)。
如果根据预测CPCA确定NDSRI等于或低于FDA推荐的AI限度,生产企业或申请人仍应制定适当的控制策略,以确保亚硝胺水平不高于AI限度。申请人应向FDA报告对已批准药物的相应变更。如果检测到的NDSRI水平超过FDA建议的AI限值,应参见第V章B节中的建议(即本文中4.2部分内容)。
4.1.2 对于正在研究和正在审评过程中的药品的推荐时间线提交前阶段:FDA建议申请人在提交首次申请之前,应对NDSRI进行风险评估,并酌情进行确证性检测。但是,如果在首次申请提交时尚未完成风险评估及确证性检测(如适用),且未提交药物主文件(DMF)变更或申请变更,上述材料可以通过修正案形式提交。此类修订应在首次申请提交后尽快提交,以尽量减少对申请审评时间线的任何潜在不良影响。
待FDA审评的申请:待审品种的申请人应尽快对产品进行风险评估,并在确证性检测发现NDSRI水平高于该指南推荐的AI限度时通知FDA。如果检测到的NDSRI水平超过推荐的AI限度,申请人应酌情修订申请。FDA将与申请人共同努力在审评周期内解决问题。
4.2 对NDSRI水平高于FDA推荐AI限度的已上市产品的建议
一般而言,对于任何药物批次,若检出NDSRI水平高于推荐的AI限度,药品生产企业不应该放行分销,并可能需要从市场上撤回药品。生产企业如果启动召回应联系FDA,FDA可根据情况行使自由裁量权,防止或缓解药品短缺。
维持上市药品供应的其他考虑如果在售药品批次中检出的NDSRI水平超过FDA推荐的AI限值,或生产企业的变更或召回有可能导致药品供应短缺,生产企业和申请人应通过邮件立即联系CDER药物短缺工作人员。若FDA被告知药品供应可能中断,FDA需根据具体问题具体分析的原则评估每种情况。FDA可能会直接与申请人沟通,考虑是否在短期内推荐临时AI限度。如果FDA推荐了临时AI限度,通常表示不反对在特定时期特定的情况下根据具体问题具体分析的原则放行含有NDSRI水平不高于临时AI限度的药品(即使该放行有可能不遵循cGMP要求)。
在特定情况下,如果多家药品生产企业被批准销售NDSRI水平不高于推荐的临时AI限值的药品,FDA将会在官网上发布推荐的临时AI限度[14]。生产企业作为责任主体,决定是否放行药品批次,并确保其药品的生产符合所有适用要求(包括cGMP),FDA希望申请人密切监控并及时向FDA报告任何用药不良反应或其他可能影响产品质量或安全的问题。
4.3 证明拟定的替代AI限度合理的方法
如果在药品中发现NDSRI水平超过FDA推荐的AI限度,FDA建议申请人采取控制措施减少或避免NDSRI产生。申请人应提交科学合理的论证依据,以证明其AI限度高于FDA推荐的AI限度的合理性。使用安全性数据可能有助于支持更高的AI限度,如获得特定化合物的数据或对替代化合物进行交叉参照评估。申请人应注意,FDA可能要求提供本指导原则之外的其他安全性数据以支持替代AI限度。如果采用特定化合物的数据或交叉参照评估方法,建议采取以下措施:
(1)致突变性评估[14-15]:当使用细菌致突变试验(Ames试验)检测NDSRI时,FDA建议采用经济合作与发展组织(OECD)指导原则471[16]中提到的全部测试菌株,采用预孵育法,使用浓度为30%的大鼠和仓鼠S9,S9通常是采用CYP450诱导剂(如苯巴比妥和β-萘黄酮组合)处理的动物而制备的。此外,应采用推荐的30 min预孵育时间作为检测致突变性信号的最佳条件。
(2)基于替代品的交叉参照评估:NDSRI可能根据替代化合物推测一个合理的AI限度,该AI限度是基于该替代化合物出现十万分之一的癌症风险确定,且可能高于根据NDSRI预测的致癌性分类而确定的AI限度。当基于替代化合物进行交叉参照评估时,申请人可根据ICH M7(R2)中所述的建议进行(Q)SAR分析,以预测潜在致突变性。FDA建议选择具有可靠致癌性数据的替代化合物,但FDA认为通常难以获得一种能充分代表NDSRI结构和机制特征且经过可靠测试的亚硝胺替代化合物。因此,由于下述化合物具有较可靠的致突变和致癌性数据,在经过合理论证的情况下应考虑将其用作替代化合物:N-亚硝基二甲胺(NDMA),N-亚硝基哌啶(NPIP),4-(甲基亚硝胺)-1-(3-吡啶基)-1-丁酮(NNK),N-亚硝基吡咯烷(NPYR)和N-亚硝基吗啉(NMOR)。
FDA说明,本节所述的证明拟定的替代AI限度合理的方法,应在实施前采用补充或修正案形式提交给FDA。
5.结语
FDA的“亚硝胺类药物相关杂质推荐可接受摄入量限度行业指导原则”,重点讨论了如何利用亚硝胺类杂质的结构特点预测其致癌潜力,并根据致癌潜力类型确定该杂质可接受摄入量限度的方法,该方法是对ICH M7(R2)中提供的方法的补充。当前国内尚未发布亚硝胺类药物相关杂质推荐可接受摄入量限度相关的指导原则,对于致突变杂质的限度要求目前国内主要参考ICH M7(R2),我国药品监管机构正在参与ICH M7亚硝胺杂质亚组对于亚硝胺杂质的国际协调工作。由于近年来国际上对N-硝基化合物的结构-活性关系的科学研究已取得一定进展,且亚硝胺类杂质常在多种药物制剂或API中发现,提供FDA对该议题及时的相关技术要求,期望能为国内含亚硝胺类杂质的药物监管和药物开发评价提供一定的借鉴和思考,降低药品的潜在致癌性风险,保障患者或受试者的健康安全。
本文仅介绍了FDA发布的《亚硝胺类药物相关杂质推荐可接受摄入量限度行业指导原则》,不代表当前国家药品监督管理局对亚硝胺类药物相关杂质的监管要求。
参考文献
[1] FDA. Recommended Acceptable Intake Limits for Nitrosamine Drug Substance-Related Impurities(NDSRIs)guidance for industry [EB/OL].(2023-08-07)[2024-12-22]. https://www.fda.gov/media/170794/download.
[2] 国家药品监督管理局药品审评中心. 化学药物中亚硝胺类杂质研究技术指导原则(试行) [EB/OL]. (2020-05-08 ) [2024-12-22]. https://www.cde.org.cn/main/news/viewInfocommon/776b663787ec5a60ac744071c3714d5a. Center for Drug Evaluation, NMPA. The guidance for
Control of Nitrosamine Impurities in Drugs [EB/OL].( 2020-05-08 ) [2024-12-22]. https://www.cde.org.cn/main/news/viewInfoCommon/776b663787ec5a60ac744071c3714d5a.
[3] FDA. Control of Nitrosamine Impurities in Human Drugs: Guidance for Industry [EB/OL].(2024-09-05) [2024-12-22]. https://www.fda.gov/regulatory-information/searchfda-guidance-documents.
[4] FDA. FDA Statement on FDA’s ongoing investigation into valsartan impurities and recalls and an update on FDA’s current findings [EB/OL].(2018-08-30) [2024-12-22].https://www.fda.gov/news-events/press-announcements/fda-statement-fdas-ongoing-investigation-valsartanimpurities-and-recalls-and-update-fdas current.
[5] ICH. M7(R2) Assessment and control of DNA reactive(mutagenic) impurities in harmaceuticals to limit potential carcinogenic risk [EB/OL]. (2023-07-25) [2024-12-
22].https://www.ich.org/page/multi-disciplinaryguidelines.
[6] Thresher A, Gosling JP, Williams R. Generation of TD50 values for carcinogenicity study data [J]. Toxicol Res, 2019, 8(5):696-703.
[7] Cross K P, Ponting D J. Developing structure-activity relationships for N-nitrosamine activity [J]. Comput Toxicol, 2021, 20: 100186.
[8] Thomas R, Tennant R E, Oliveira AA F, et al. What makes a potent nitrosamine? Statistical validation of expertderived structure-activity relationships [J]. Chem Res Toxicol, 2022, 35(11): 1997–2013.
[9] Ponting D J, Dobo K L, Kenyon M O, et al. Strategies for assessing acceptable intakes for novel N-nitrosamines derived from active pharmaceutical ingredients [J]. J Med Chem, 2022, 65: 15584-15607.
[10] Li Y, Hecht S S. Metabolic activation and DNAinteractions of carcinogenic N-nitrosamines to which humans are commonly exposed [J]. Int J Mol Sci, 2022,23(9): 4559.
[11] Thresher A, Foster R, Ponting D J, et al. Are all nitrosamines concerning? A review of mutagenicity and carcinogenicity data [J]. Regul Toxicol Pharmacol, 2020,
116: 104749.
[12] FDA. Recommended Acceptable Intake Limits for Nitrosamine Drug Substance-Related Impurities (NDSRIs) [EB/OL].(2024-11-20) [2024-12-22].https://www.fda.gov/regulatory-information/search-fdaguidance-documents/cder-nitrosamine-impurityacceptable-intake- limits.
[13] FDA. Updates on possible mitigation strategies to reduce the risk of nitrosamine drug substance-related impurities in drug products [EB/OL].(2021-11-18) [2024-12-22].https://www.fda.gov/drugs/drug-safety-and-availability/updates-possible-mitigation-strategies-reduce-risknitrosamine-drug-substance-related-impurities.
[14] FDA. CDER Nitrosamine Impurity Acceptable Intake Limits[EB/OL]. (2024-11-20) [2024-12-22]. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/cder-nitrosamine-impurity-acceptable-intake-limits.
[15] Li X, Le Y. Revisiting the mutagenicity and genotoxicity of N-nitroso propranolol in bacterial and human in vitro assays [J]. Regul Toxicol Pharmacol, 2023, 141: 105410.
[16] OECD. Test No. 471 Bacterial Reverse Mutation Test:OECD Guidelines for the Testing of Chemicals, Section 4[EB/OL]. (2020-07-29) [2024-12-22]. https://www.oecd.
org/en/publications/test-no-471-bacterial-reverse-mutationtest_9789264071247-en.html

来源:中国知网
关键词: 亚硝胺