
嘉峪检测网 2025-06-22 18:39
导读:本文通过对EMA 和FDA 发布的2 个重要文件进行总结,以及化学药品、抗体类生物制品和抗体偶联药物申报的实际案例分析,说明灵活性并非等同于减少药学研究和(或)需要递交的数据,而是通过科学工具和策略实现临床获益与风险的平衡。创新药全球同步递交的进程才刚刚开始,期望本文能为我国加速药学研发技术指导原则的研究制定及后续落地实施提供参考,助力解决药学研发
摘 要Abstract
为助力创新药品的加速研发,解决未被满足的医疗需求,中国、美国和欧盟的药品注册管理法规均设立了加速申报途径,提高了公众用药的可及性和应对突发公共卫生事件时医药供应的保障能力和管理效率。但相关产品的药学研发仍面临不能在有限时间内完成满足传统新药上市申请需要的全部研究和数据等挑战。为此,美国食品药品监督管理局(FDA)和欧洲药品管理局(EMA)与工业界开展了系列研讨,并发布了一系列指南和工具等文件,为加速申报途径产品的药学研发提供了灵活性。例如,通过先验知识、风险评估、创新的工艺验证策略等工具,将部分研究和数据的递交后置,并结合批准后变更管理方案(PACMP)等上市后变更工具,在控制风险的同时,可显著加快有临床价值的创新药物上市。本文通过对EMA 和FDA 发布的2 个重要文件进行总结,以及化学药品、抗体类生物制品和抗体偶联药物申报的实际案例分析,说明灵活性并非等同于减少药学研究和(或)需要递交的数据,而是通过科学工具和策略实现临床获益与风险的平衡。创新药全球同步递交的进程才刚刚开始,期望本文能为我国加速药学研发技术指导原则的研究制定及后续落地实施提供参考,助力解决药学研发和注册环节等方面的挑战,进而推动全球创新药物在我国实现同步研发和上市。
To accelerate the development of innovative medicines and address unmet medical needs, regulatory authorities in China, the United States, and the European Union have established expedited pathways. These programs have enhanced public access to medicines, ensured emergency responses, and improved efficiency of health authorities. However,products under accelerated pathways often face challenges in completing the full scope of Chemistry, Manufacturing,and Controls (CMC) studies and data requirements within constrained timelines. In response, the U.S. Food and Drug Administration (FDA) and European Medicines Agency (EMA) have collaborated with industry to develop flexible regulatory tools, publishing key guidelines that offer flexibilities such as strategic deferral of certain studies or data submissions.These tools, including but not limited to prior knowledge, risk assessment, innovative process validation strategies, and Post-Approval Change Management Protocols (PACMPs), enable earlier market access while ensuring product quality and managing residual risks. This article reviews two important guidelines from the EMA and FDA and presents case studies involving small molecules, biologics (antibodies), and antibody-drug conjugates. It illustrates that regulatory flexibility does not mean reducing the scope of CMC studies or data packages for submission, but rather applying science-based tools to balance product quality risks and clinical benefits. The journey of China joining globally simultaneous submissions of innovative drugs has only just begun. We hope this article offers insights to inform the development of technical guidelines for accelerated pharmaceutical programs, supporting earlier access to innovative medicines for patients in China.
关键词Key words
加速路径;工艺验证;先验知识;药学研究;风险评估;药学;全球同步递交
accelerated pathways; process validation; prior knowledge; pharmaceutical development; risk assessment;chemistry, manufacturing and controls (CMC); global simultaneous submission

为了助力创新药品的加速研发,解决未被满足的医疗需求,中国、美国和欧盟的药品注册管理法规均设立了加速申报途径。相关政策和举措不仅能促进医药产业的发展,还有助于提高公众用药的可及性和应对突发公共卫生事件时医药供应的保障能力和管理效率。
在欧盟,现行的加速审评审批途径包括加速审评(accelerated assessment)、附条件上市许可(conditional approval)、优先药物(priority medicines,PRIME) 通道和适应性路径(adaptive pathways)。美国食品药品监督管理局(Food and Drug Administration,FDA)制定并成功推行了4 种不同的路径以加快新药研发和上市注册审评审批, 包括突破性疗法(breakthrough therapy,BT)、优先审评(priority review,PR)、快速通道(fast track,FT) 和加速批准(accelerated approval)。
我国2020 年修订的《药品注册管理办法》为新药上市开辟了4 条“ 快速通道”,即突破性治疗药物程序、附条件批准程序、优先审评审批程序和特别审批程序,这一里程碑式的改革举措,极大地激励了国内外制药企业研发创新的热情,对加快创新药物上市产生了积极影响。
在创新药物的研发进程中,企业需要确保临床研究、非临床研究和药学研究均符合相关要求。快速的适应性临床研究是加速创新药研发的关键途径之一,但这一举措会缩短产品研发周期,进而导致药学研究成为影响产品研发进程的关键因素。因此,在确保产品质量和患者安全的前提下,企业利用先验知识、加速模型和工具等制定有效的研发策略,加快药学研发,使患者尽早获得创新药物,成为业界普遍关注和热议的话题。尤为重要的是,企业通常需要就研发策略和计划,及时与药品监管部门进行沟通和讨论,以便针对药学研发所需的数据达成共识。本文通过介绍并总结欧洲药品管理局(European Medicines Agency,EMA) 和FDA 发布的关于加速药学研发的文件,结合实际案例分析,从药学角度出发,对我国具有明显临床价值的创新药品如何更快惠及患者进行思考与展望,以期为我国加速药学研发技术指导原则的研究制定及后续落地实施提供参考,助力解决药学研发和注册环节等方面的挑战,进而推动全球创新药物在我国实现同步研发和上市。
1 EMA 与FDA 加速审评审批药物的药学研发工具和指南演化
自2014 年EMA 推出适应性路径程序后,国际药品监管机构开始聚焦于如何加速将科学突破转化为新的高质量产品。2016年,EMA 进一步推出PRIME通道。然而,在当时,对于这些加速审批路径, 在药学研发化学、生产和控制(chemistry,manufacturing and controls,CMC)方面的讨论相对有限。
2015 年1 月, 欧盟制药工业协会(European Federation of Pharmaceutical Industries and Associations,EFPIA)、欧洲生物制药企业(European Biopharmaceutical Enterprises,EBE)与EMA 就药学关键技术和监管进行了研讨,并于2017 年12 月完成了关于加快药学研发的EFPIA-EBE 白皮书定稿[1]。该白皮书聚焦于潜在可满足未满足的医疗需求的药物,旨在探索既能保障药品质量与安全,又能被监管机构认可的可选择的加速药学研发方案。书中重点剖析了加速药学研发面临的挑战及基本要素,并详细阐述了制药行业为实现加速申报路径产品尽早上市而采取的药学研发实践策略。
2017 年11 月, EMA 生物制品工作组(Biologics Working Party,BWP) 和质量工作组(Quality Working Party,QWP)召开了关于先验知识的研讨会,探讨了先验知识的含义以及如何用于支持产品研发、制造和控制策略,并通过一些具体的行业实际案例探讨了加速药物上市所面临的药学挑战[2]。
2018 年 EMA、FDA 联合工业界等利益相关方,举办了关于支持加速可及通道的药学研发方法的研讨会[3],提出及时研发和向患者提供高质量药物以满足未满足的医疗需求,是监管机构和工业界的源动力;讨论了药品加速研发和早期准入过程中可能出现的药学挑战;强调通过结合实际案例研究,加强工业界和监管机构之间的协作和沟通;提出利用新兴的科学知识及多样化的监管途径,在确保药品质量且不损害其安全性和有效性的基础上,助力患者及时获得创新药。
2022 年4 月,EMA 发布了《针对未满足临床需求的 PRIME和特定上市申请的质量数据包的科学要素和监管工具的工具箱指南》(Toolbox Guidance on Scientific Elements and Regulatory Tools to Support Quality Data Packages for PRIME and Certain Marketing Authorisation Applications Targeting an Unmet Medical Need,以下简称《工具箱指南》)[4]。该指南系统总结了欧盟现有监管框架内可用的科学要素和监管工具,可用于支持药物研发和完成模块3 质量数据包,为指定的PRIME 药品上市许可申请(marketing authorization applications,MAA)和针对未满足医疗需求的部分MAA 申请提供支撑。同年,FDA 发布了《加速项目产品质量评估》(MAPP5015.13 Quality Assessment for Products in Expedited Programs)政策和程序手册(manual of policies and procedures ,MAPP)[5],供其药品质量办公室(Office of Pharmaceutical Quality,OPQ) 使用。其目的是促进和加快新药上市申请(new drug application,NDA) 和生物制品上市许可申请(biologics license applications,BLA)的研发、评估和上市审评审批流程,以解决严重或危及生命的疾病治疗中未满足的医疗需求。该MAPP 为临床研发加速的产品应对药学挑战提供了指导,促进了BT、FT 途径产品,以及《处方药使用者付费法案》(Prescription Drug User Fee Act, PDUFA VII)承诺书中包含的其他相关产品的药学研发。该MAPP 还概述了OPQ 如何运用《美国联邦法规》(Code of Federal Regulations,CFR) 第21 篇( 以下简称21CFR)314.105 中预期的监管灵活性, 帮助克服药学数据包难以满足传统监管要求的难题,从而使患者能够更早地获得这些产品。
2023 年,EMA 和FDA联合举办了一场研讨会,FDA下属机构药品审评与研究中心(Center for Drug Evaluation and Research,CDER)与EMA联合发布了关于PRIME 和BT申请的质量和药品生产质量管理规范(good manufacturing practices,GMP) 的问答文件(EMA–FDA Joint Q&As on Quality and GMP Aspects of PRIME/Breakthrough Therapy Applications,以下简称《问答》)[6],该文件侧重从实际应用层面为申请人加速药学研发提供指导。
1.1 EMA《工具箱指南》简介
EMA 在2022 年4 月发布的《工具箱指南》中所提到的工具是指用于药品研发、制造和质量风险管理的科学概念、原理或技术。该指南针对药品临床使用至关重要的质量问题和(或)属性进行了阐述,包括先验知识、风险评估、工艺验证、标准制定、GMP 合规性、稳定性和可比性以及早期识别等。通过运用这些概念、原理或技术,可在递交部分CMC 数据时提供一定的灵活性,但需要注意的是,这并不等同于可以降低支持上市申报资料的质量,产品的质量、安全性和有效性仍需要得到充分证明。此外,为充分发挥这些监管工具的作用,EMA强烈建议申请人尽早与监管机构展开对话,共同讨论其总体开发计划,包括质量计划和供应链合规性等,以确保双方就数据要求达成一致意见。以下将简要介绍《工具箱指南》中列出的部分工具。
1.1.1 先验知识
先验知识是国际人用药品注册技术协调会(International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use,ICH)指南(如 Q8、Q10和 Q11 等),以及FDA 和 EMA所发布相关指南中使用的术语。先验知识涵盖多个方面,既包括从研发和制造实践中积累的经验知识(如基于类似化合物、产品和工艺所获得的经验),也涉及对科学文献的参考,或是已确立的科学原理的应用。
如果先验知识被证明适用于在研产品,则可基于先验知识来调整部分质量研究工作的完成时间节点,或者使用先验知识替代某些在研产品的质量研究数据(如稳定性研究、工艺验证、质量标准论证等数据)。但如果先验知识与在研产品的经验无关,而是基于类似产品,则需要证明该先验知识对在研产品的适用性。
此外,先验知识也可以源自“平台”,例如,单克隆抗体平台产品通常使用相似的配方、制造工艺和(或)分析测试方法,这些相关经验与方法构成了先验知识的一部分。
1.1.2 风险评估
ICH Q8 、 Q9 中提到,风险评估是一种基于科学的系统化流程,用于整合相关信息,以支持风险管理流程中的风险决策。该流程包括识别危害以及对与接触这些危害相关的风险的分析和评估。在药物研发过程中,风险评估是一项常用的工具,用于评估药物配方和制造过程,了解材料属性和工艺参数对产品质量的影响,定义其关键性并为后续研究提供参考依据。此外,风险评估还可用于评估申报资料要素,如关键属性、模型的适用性和先验知识等,并为总体控制策略提供信息支持。申请人应基于已识别的风险概况来证明上市许可申报资料各部分中可用数据的范围。
值得注意的是,风险评估自产品研发之初便已启动,并随着对产品及其特性的了解增加而逐渐成熟。监管机构应基于产品的获益- 风险评估,分析产品使用替代支持数据替换某些常规数据可能产生的潜在风险。当然,基于风险的方法同样适用于其他具有临床价值的产品,尤其是先进疗法类产品(advanced therapy medicinal product,ATMP)。
1.1.3 工艺验证
适用于加速研发的工艺验证方式包括同步(concurrent)验证、解耦(decoupling)验证(原料药/ 原液和制剂的验证解耦)、连续工艺确证,以及在提交NDA时以工艺验证方案代替实际的验证活动。这些验证方式具有多种优势:①可以通过减少递交时的工艺验证批次、借助先验知识以及来自非工艺性能确认(process performance qualification,PPQ)批次或小规模批次的验证数据来支持工艺性能验证。②在提交NDA 时,以工艺验证方案来替代某些工艺验证活动,如树脂寿命研究、新细胞库的引入、新标准物质的引入、保持时间研究、运输验证、再处理等。③为了避免成品工艺验证活动出现延迟,可以使用在正式工艺验证之前生产的原料药/ 原液批次来开展成品工艺验证。需要注意的是,这些原料药/ 原液批次必须是在GMP 受控条件下严格按照注册工艺生产的,且应该证明其能够充分代表商业制造工艺, 并满足相应的预期标准。④ 当对特定生产工艺拥有大量先验知识,并且生产过程中包含广泛的在线控制时, 可以使用连续工艺确证来验证生产工艺, 从而促进快速研发。
1.1.4 质量控制策略
加速研发在质量控制策略方面会面临许多挑战,例如,生产和临床经验相对匮乏、生产批次有限导致难以充分评估工艺的一致性,工艺和方法验证研究尚未完成,对关键属性以及工艺与质量之间的内在联系的理解不够深入等。因此,与采用标准研发流程的产品相比,加速申报途径的产品在获批时,用于支持质量控制策略的数据量可能相对较少。为了应对这些挑战,申请人可以通过增加质量标准中的检测项目、强化过程控制、增加工艺参数、确定更多关键工艺参数、设置更窄的关键工艺参数范围等方式,抵消由于加速研发而导致的产品和工艺知识水平下降所带来的风险。待产品上市后,随着更多数据的积累,申请人可以通过变更程序更新控制策略,在此过程中, 也可以使用ICH Q12 工具进行变更,如批准后变更管理方案(post-approval change management protocol,PACMP)。
对于质量标准,其可接受标准和限度可以比临床研究批次的放行标准更为宽松。此外,在建立标准时,通常还会考虑临床经验以外的其他信息来源,如体外研究数据、动物实验数据、已发表的参考文献、基于特定研发平台的先验知识以及关键质量属性的潜在影响等。关键质量属性标准限度的合理性应与临床实际表现相关联,而不应仅仅基于容差区间等统计方法得出。仅对有限批次进行统计分析,可能会导致标准限度过于宽泛,无法从临床角度证实其合理性。
1.1.5 稳定性研究
在加速研发计划中,可能无法获得标准的稳定性数据包,如稳定性研究批次少于3 个批次或者研究时长较短,在这种情况下可以使用替代方法来证明产品的稳定性。以加速研发路径下的生物制品为例,可以使用根据结构相似分子稳定性先验知识构建的稳定性模型来推断稳定性数据的变化趋势,从而预测产品的效期。而对于小分子产品,则可进行稳定性外推等操作。
1.1.6 可比性研究(生物制品)
可以使用基于风险的方法来设计可比性研究。对于加速研发的产品,基于风险评估结果,可比性研究可聚焦于相关的关键质量属性,或者使用先验知识来预测生产相关变更可能产生的影响。
1.1.7 监管工具
对于加速研发的产品,在其研发阶段,EMA 会通过提供科学咨询服务的方式对研发活动提供指导,并通过提前介入以及与申请人保持及时的沟通交流,针对产品研发过程中遇到的问题提前进行讨论并达成一致意见。EMA的法规中也规定了加速审评方法,以缩短审评时限。EMA 鼓励申请人主动与EMA 沟通,讨论加速审评以及与申报资料相关的任何问题。由于加速研发可能会影响到GMP 合规水平以及GMP检查范围, 因此申请人也应就GMP 相关问题及早与EMA 进行讨论并达成一致意见,确保在提交NDA 时生产场地已做好接受检查的准备,避免因此影响审评时间。此外,申请人也可以借助PACMP 合理规划上市后的变更事宜。
1.2 FDA《问答》简介
EMA 正式发布《工具箱指南》的同年,FDA 发布了《加速项目产品质量评估》[5],旨在为BT、FT 等途径产品的加速药学研发提供指导。2023 年, CDER与EMA 联合发布了《问答》[6]。作为《工具箱指南》的补充,《问答》回答了对《工具箱指南》中各工具在药物研发和审评实际应用中的问题。对EMA 来说,《问答》适用于所有药品种类, 包括复杂生物制品(如ATMP)。而对FDA 而言,《问答》对 21 CFR314.105 (c) 中提到的“ 监管灵活性” 的概念作出了更为具体的阐释, 且与此前发布的其他指南一样,均体现了FDA 在评估产品质量风险及获益方面的理念;与MAPP 相同,《问答》适用于CDER 监管的药品和生物制品, 而不适用于生物制品审评与研究中心(Center for Biologics Evaluation and Research,CBER)监管的产品,如ATMP。《问答》以4 个附件的形式对一般原则进行了详细阐释,具体内容如下。
1.2.1 PRIME/BT 申请控制策略注意事项问答
由于PRIME/BT 申请具有加速特性,药品在临床使用方面的经验和对产品质量和工艺的了解相对有限,因此,在设定质量标准时,通常只能基于少量的研发批次和商业批次数据。在这种情况下,除临床经验外,还可以使用其他数据来支持质量标准的建立,包括使用动物实验数据、体外研究数据以及基于特定研发平台的先验知识等。其中,先验知识还可支持整体控制策略的建立,包括关键质量属性、关键工艺参数的确定,以及工艺参数范围的设定。在确保临床安全性和有效性的基础上,质量标准有可能设定得比临床批次的测试结果更为宽松。而整体控制策略则有时需要调整,例如,设置更窄的工艺参数范围、识别额外的关键质量属性和(或)关键工艺参数,以及在质量标准中纳入额外的工艺控制或检测项目。随着研究的持续开展,申请人可提前计划,在产品上市后基于更多数据修改质量标准和(或)控制策略,并与监管机构沟通,通过PACMP 进行递交。需要注意的是,PRIME/BT 产品的分析方法验证还应符合ICH Q2 (R2)[7] 和 Q14[8] 的相关原则。
1.2.2 PRIME/BT 申请工艺验证方法问答
EMA 和FDA 对验证过程的监管略有不同,特别是化学药品和生物制品。对于按标准工艺生产的化学药品,EMA 仅要求在上市申报资料中包含工艺验证方案,而无需提供商业规模的工艺验证数据。商业规模的工艺验证应在药品发运分销之前完成。而FDA要求进行3 个阶段的工艺验证[9],即化学药品的上市申报资料中需包括工艺验证的第一阶段工艺设计,并在药品发运分销之前完成第二阶段PPQ。第三阶段持续工艺确认则从完成PPQ 后开始,贯穿药品剩余生命周期。对于生物制品,在通常情况下,EMA 和FDA均要求在上市申报资料中包含商业规模的工艺验证(PPQ)数据。
对于患者的获益- 风险比很高的药品,包括用于防治重大疾病的药品、有效期极短的药品(如放射性药品)和临床急需药品等,可采用同步验证。欧盟 GMP 指南附件 15 [10] 将同步验证定义为在特殊情况下,以患者的高收益风险平衡为基础的验证,验证批可作为商业化批。我国GMP 也提到了同步验证[11]。同步验证方案应递交给监管机构,方案中需包括有关验证活动、批次数量、检测项目和放行标准等的详细信息。工艺研发阶段积累的知识和数据、先验知识和小规模工艺生产的数据可用于支持同步验证。这些数据应能够证明工艺过程处于受控状态, 并能持续制造出符合质量标准的药品。在NDA获批后, 按批准条件生产的批次药品若符合拟定的质量标准,通常不需要监管机构批准即可放行。但同步工艺验证数据应在获批后提交给监管机构。若验证活动或结果不符合验证方案,企业需进行充分的根本原因调查, 并与监管机构共同讨论失败的性质、调查结果以及对产品质量的影响, 以决定是否将该批次药品投放市场。
通常情况下,原料药/ 原液的验证批次用于生产制剂的验证批次。在某些情况下,也可以接受将制剂的工艺验证与原料药/ 原液的工艺验证分开进行,如使用临床批次或符合GMP 的研发批次来生产制剂验证批次。这种方法的适用性取决于用于制剂验证的原料药/ 原液批次是否具有代表性。该方法不仅适用于PRIME/BT 产品,也可能适用于其他情况。《问答》推荐相关企业事先与监管机构讨论该方法的适用性。
1.2.3 PRIME/BT 申请确定复测期或有效期替代方案问答
在EMA 和FDA 同意的前提下,可以接受在NDA 中递交低于ICH Q1A(R2)[12] 和Q5C[13]要求的稳定性数据。在可比性得到证实的情况下,允许使用其他批次的长期稳定性数据代替稳定性批次的稳定性数据,以制定复测期或有效期。在获批后,申请人应继续按照批准的稳定性方案进行稳定性研究,并根据要求按照与相关监管机构达成的协议提交数据,以确认复测期或有效期。如果申请人能够提供足够信息证明数据的相关性,先验知识也可用于支持复测期或有效期的制定。
对于化学药品,FDA 和EMA均允许使用经过验证的模型来制定复测期或有效期。申请人需提供充足的信息来支持模型的使用。FDA 要求最晚在NDA 前会议(pre-NDA meeting)时递交使用模型的申请,EMA 则要求在PRIME 启动会上提出申请,并通过科学建议(scientific advice)跟进。
FDA 通常不推荐对CDER监管的生物制品使用稳定性模型制定复测期或有效期,仅在支持证据充分的情况下予以考虑。
1.2.4 PRIME/BT 申请 GMP注意事项问答
在满足特定前提条件的情况下,临床试验生产场地也可用于商业化生产。EMA 和FDA 会基于各自的法规,基于风险启动核查。在加速审评流程中,申请人应及时提交信息、制定计划,确保审评和核查同步进行。在部分特殊情况下,监管机构可能会同意申请人使用在适当的GMP 级别下制造的试验药品的起始物料,也可能允许将为关键临床研究生产的现有批次进行商业化。
2 EMA 和FDA 审评中应用加速药学研发工具的案例分析
2.1 化学药品案例1
某创新化学药在针对某一肿瘤适应症的治疗中表现出明显的临床疗效。在申请人与FDA 进行沟通交流时,FDA 建议申请人申请BT 审评路径,以缩短审评周期。但如果按原定的药学研发计划,通过公司标准的验证流程开展验证活动,即便获得FDA 批准,药品仍需要 在4 个月后才能提供给患者使用。为使患者能尽早使用到这款创新药品,申请人与 FDA 展开讨论,并提出如下方案:将原料药和制剂的验证脱离(即解耦验证),使用临床批次的原料药进行3 批制剂工艺验证,其中1 批验证批将用于上市销售。同时,申请人承诺在常规制剂商业化生产之前,用验证批所使用的原料药进行3 批制剂验证(即对前3 批商业化制剂生产进行相当于PPQ 验证批的高取样和检测频率),并提供相应的报告。
为支持该方案, 申请人向FDA 提供了多批次历史数据,以证明临床批次与拟商业化批次的制造工艺和质量具有可比性。在现场核查期间,FDA 的核查员、审评员与申请人内部专家和质量部门之间进行了密切的沟通交流,各方就风险评估达成了平衡且一致的看法。最重要的一点在于,在整个过程中,各方始终聚焦于确保患者能够及时获得创新药品。最终,FDA 接受了申请人的提议,且通过现场核查认定申请人满足相关条件,患者得以在药品获批后第一时间使用到这款创新药品。
2.2 化学药品案例2
2021 年, 美国FDA 批准了一款小分子药物。该药物的NDA 审评工作在FDA 肿瘤卓越中心(Oncology Center of Excellence,OCE) 所倡议的Orbis 项目合作框架下进行。Orbis 项目为肿瘤药物在国际合作成员国家和地区的同步申报和审评提供了框架支持。此次审评由美国FDA 与澳大利亚治疗用品管理局(Therapeutic Goods Administration,TGA)、加拿大卫生部(Health Canada,HC)、英国药品和健康产品管理局(Medicines and Healthcare Products Regulatory Agency,MHRA)合作进行。在Orbis 项目合作框架下,各成员国家和地区能够协作审评、互信共享,从而加快批准速度(较FDA 设定的目标日期提前了约1 个月)。该药物的适应症申请被授予PR 资格,且该药物分子还获得了FDA 罕见病用药认定和BT 认定,这意味着允许企业使用替代验证策略,且企业拥有更多与FDA 沟通交流的机会。
在此案例中,加速审评在药学方面的关键促成因素之一是采用了同步验证策略,即在其余工艺验证批次尚未完成的情况下,首个工艺验证批次就已获批放行并在美国上市销售。后续工艺验证批次则逐步完成验证工作,用于支持临床研究和药品商业上市的供应。该同步验证策略以及与药品监管机构达成的共识助力加速药品上市,并避免了资源的大量浪费。同时,企业针对其他技术问题,主动与药品监管机构进行讨论并达成共识,包括起始物料的选择、M7 致突变杂质控制策略、稳定性数据的提交和药学资料的滚动提交等。
2.3 新冠治疗用生物制品案例
在新冠疫情大流行期间,为了加快一款治疗新型冠状病毒感染(COVID-19)的单克隆抗体药物的研发,申请人与EMA 和FDA 进行了多轮沟通。双方就如何基于科学和风险的方法,以及如何最有效地利用现有监管工具(如承诺、方案)等方面达成一致意见。对于该药物而言,能够尽早进入市场供应的关键在于获得临时或紧急使用授权,及时安排产品供应,并制定提交完整的MAA 计划。
例如,基于原液所有临床批次的总体实践经验,监管机构同意将原液和制剂的工艺验证解耦。同时,在制剂的第二阶段PPQ 中,可使用一批临床批次的原液以加速工艺验证进度,并允许审评期间滚动提交工艺验证报告。在质量控制方面,基于先验知识和临床相关数据,监管机构同意在初始市场供应中采用相对宽松的质量标准,且申请人承诺会在获得更多生产经验后,对质量标准进行审核和必要的修改。此外,最初递交的资料中仅包括分析方法确认(而非验证)数据,申请人承诺在提交完整MAA 时会补充提供分析方法验证数据。在临床阶段和初始市场供应阶段,仅使用酶联免疫吸附分析(enzyme linked immuno sorbent assay,ELISA)进行效价测定,申请人承诺在关键临床试验结束后,采用细胞法生物活性检测进行效价测定。
为加速生产进程,监管机构同意在进口原液批次的放行检测完成前,将该批次原液投入制剂生产。对于供应至欧盟的产品,由于该产品生产商所在国家未与欧盟达成互认协议,通常应在进口时进行放行检测。为加快药物的市场供应,EMA 豁免了3 个月的进口检测,允许在欧盟以外国家和地区进行产品检测和放行工作。
在申请人与EMA、FDA的早期交流中,各方均认可应快速推进药物的研发和临床试验项目, 并且考虑到未来为满足供应需求, 需要快速扩大工艺规模。这意味着在关键临床研究期间或之后, 以及早期上市后维护阶段,将进行重大变更。该单克隆抗体产品在临床研究完成后及上市后维护早期阶段,引入了多个重大药学变更, 包括细胞库建立、生产场地变更和工艺规模变更(表1)。大部分变更在符合监管机构同意的方案和承诺等的相关要求后可立即予以实施,而无需在提交完整的数据包后等待监管机构的审评审批。表1 中所述变更的工艺和分析可比性,通常基于少于3个批次的数据。可比性的确定主要依据原液批次的分析数据和稳定性数据,而无需制剂批次数据或体内研究数据。由于已证明这些变更不会对产品质量和稳定性产生负面影响,使用非单克隆细胞来源生产的临床批次的外推数据成功将药品的有效期从12 个月延长至18 个月。
表1 某新冠治疗用生物制品从临床阶段至长期商业化供应期间发生的重大药学变更
| 项目 | 临床阶段 | 新药上市申请和早期商业化供应 | 长期商业化供应 |
|---|---|---|---|
| 细胞来源 | 非单克隆细胞 | 单克隆 MCB | 新 WCB |
| 原液生产商 | 国家 1 的 CMO | 国家 1 的 CMO | 国家 2 的 CMO |
| 制剂生产商 | 国家 A | 国家 B | 国家 B |
| 规模(L) | 2000 | 12 000 | 15 000 |
| 设备 | 一次性系统 | 一次性系统 | 不锈钢设备 |
| 效价检测方法 | ELISA | 细胞法生物活性检测 | 细胞法生物活性检测 |
2.4 治疗用生物制品案例
此案例涉及一款2014 年获得美国FDA 批准的治疗用生物制品。当时,该生物制品虽处于I 期临床试验阶段,但已获得了FDA的BT 认定。为了加快药学研发的准备工作,申请人采取了一些灵活性措施。
工艺验证解耦策略。原液工艺验证与制剂工艺验证采用解耦策略,即通过确保进行工艺验证的原液与用于制剂工艺验证的原液在生产过程中无重大工艺变更,来实现原液工艺验证和制剂工艺验证的同步完成。在研发时间线中,并行开展原液和制剂工艺验证可节省4~6 个月的时间,且不会对药品的质量、安全性和有效性造成额外风险。这种情况下,工艺验证就不再是药学资料提交的限速因素。
原液生产场地申报策略。为了满足商业化和临床需求,在美国申报并获批的初始BLA 中包含2 个原液场地。在提交的药学资料中,纳入了原始临床批次场地和新场地之间分析的可比性数据,以支持审评审批。此外,申请人与FDA 就2 个原液生产场地的可比性研究方法和提交内容进行了多次沟通。
基于分析可比性的剂型变更。申请人与FDA 进行密切沟通,及时讨论并就可接受的注册审评路径达成一致意见,对该产品药学研发的成功至关重要。例如,剂型的供应策略发生了变更,即从输注用冻干粉转换为液体小瓶。就此策略更新,申请人提前与FDA 进行了充分研讨,最终基于分析可比性的结果,将液体小瓶剂型作为上市后变更进行申报并获得批准。
基于平台数据的宿主细胞蛋白(host cell protein,HCP)测定方法灵活性。在该产品上市申报时,工艺或产品特定HCP测定方法尚未完成研发。如果等待该方法研发完成,将导致上市申报周期延迟6~9 个月。为了在药学研发方面实现加速,企业提交了市售HCP 测定方法。考虑到企业的平台数据已证实工艺中具备相应的清除率,FDA 批准将市售HCP 测定方法作为初始商业放行方法,并在BLA 审评期间就工艺或产品特定HCP 测定方法的研发达成一致,将其作为上市后承诺事项。上述举措在确保临时解决方案不会对患者的安全性和有效性造成任何风险的前提下,避免了工艺或产品特定HCP测定方法成为药品获批上市的阻碍。
2.5 抗体偶联药物案例
为加速抗体偶联药物(antibody-drug conjugate,ADC)的上市进程,公司在商业化规模生产PPQ 环节采用了解耦验证策略。该策略将抗体中间体、原液和制剂的PPQ 分开进行:使用PPQ 前的抗体中间体(商业化生产规模GMP 批次)来验证原液的生产工艺;使用PPQ前的原液(商业化生产规模GMP批次)验证制剂的生产工艺。同时,确保PPQ 前后批次在生产规模、设备及参数等方面的一致性,以保证药学质量的可比性和连贯性。为满足监管要求,公司在提交BLA 前,与FDA 进行了充分沟通并达成一致:提交的资料须至少包含1 批使用充分验证的原液(即使用抗体中间体PPQ批次生产的原液)生产的制剂批次数据,且这些数据需涵盖批放行信息及过程控制信息。
解耦策略使抗体中间体、原液和制剂工艺验证能够并行推进,从而显著缩短验证周期,加快研发进度,同时确保数据的科学性和可靠性。
在商业产品货架期设定方面,FDA 认可工艺代表性批次的实时稳定性数据,这些数据可来自非商业生产厂批次。这一方法强调了具有工艺代表性的、非商业生产厂批次的稳定性数据在商业产品货架期设定中的重要性,符合ICH Q1A 及Q5C 所体现的相关原则,使得企业能够凭借有限的商业生产厂批次稳定性数据,获得更长的商业产品货架期批准。
3 抗体偶联药物药学监管考量的文献案例分析
2023 年9 月,Journal of Pharmaceutical Sciences 发表了一篇文章《抗体偶联药物药学监管考量》(CMC Regulatory Considerations for Antibody-Drug Conjugates)[14]。该文章汇聚了全球多家领先制药企业在ADC产品质量控制策略、可比性研究、工艺验证策略和处方研发等方面的多维度见解,文中提到了解耦验证策略( 图1)。
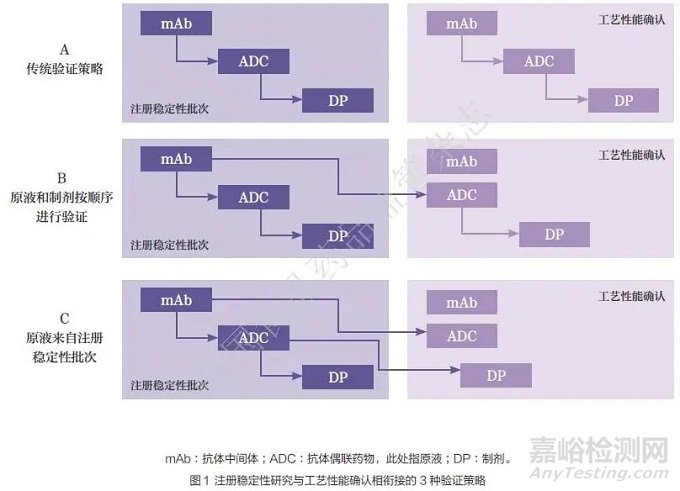
工艺验证的核心目标在于,通过确凿的证据表明生产工艺流程能够稳定、可控地运作,并持续产出符合既定质量标准的产品。基于这一核心目标提出的解耦验证策略,是利用具有工艺代表性的批次,分步骤开展抗体中间体、原液和制剂等的工艺验证工作。此策略不仅可以实现时间、原材料及成本的节约,而且还允许通过多个具有生产代表性的批次,全面覆盖并验证整个工艺流程,从而确保验证工作的全面性和有效性。
4 展望与结语
近年来,随着医药科学技术的不断演进、全球药品监管机构对未满足的临床需求的日趋重视,以及以患者为中心的理念逐渐深入人心,研发人员与监管机构开始共同探讨如何更好地以临床需求为导向,在控制风险的同时,更快速地使治疗方法和产品惠及患者。其中,在法规层面,建立多种快速通道以支持加速研发和加速审批是一项重要的监管举措。但同时,工业界和监管机构在实践中观察到,加速研发的产品极有可能面临CMC 数据要求的挑战。由于研究批次更少、研发周期缩短等原因,这些产品可能无法产生完整的CMC 数据包。为了解决这些问题,欧美等监管机构陆续出台了相关指南及问答文件等,以明确在临床获益大于风险的前提下,如何为快速通道产品的CMC 要求提供灵活性。这些监管举措的实施,确实取得了显著成效。基于本文案例中企业在美国和欧盟的申报经验,药学加速工具的应用能够使患者提前4~9 个月获得创新药物。
在我国药品审评审批制度深化改革的进程中,应进一步加大鼓励创新力度,推动国际通行药品监管规则在国内的转化落地实施,加速全球创新药物在我国的同步研发,促进其同步申报、审评和上市。目前,我国4 条快速审批路径等政策的实施,已助力越来越多的全球创新药、儿童用药及罕见病用药等更早地在我国上市。然而,创新药在我国和其他国家及地区的同步递交进程才刚刚开始,且大多为新适应症上市申请,申请人可以使用已有的完整的药学资料支持在我国的上市递交和批准。随着我国支持同步研发政策的推进,未来将有越来越多的创新药品的首个上市申请在我国和其他国家及地区同步递交。但需要注意的是,在药学研发和注册方面依然面临很多挑战,例如,在全球一体化临床和药学研发策略布局下,加速研发产品在NDA 时已有的药学研究数据无法满足我国传统上市申请药学资料要求(包括完整的原液/原料药、制剂工艺验证报告,注册检验商业化样品批次及数量,以及足够的长期稳定性数据以支持必要的药品货架期等)。此外,如何进行复杂的上市后药学变更管理,以确保创新药品的持续供应等,也是我国药品监管部门和制药行业亟待解决的问题。本文总结了欧美监管机构和行业的观点与经验,介绍了相关指南形成历史和实际案例经验,包括:重视先验知识的使用,在递交时应用先验知识(包括平台知识)设置临床相关的质量标准,包括有效期等;在药品获批后,随着更多数据和知识的积累,可根据需要通过PACMP 或者其他法规工具修订质量标准和延长有效期;在接受一定风险的前提下,保证获批时的产品质量;企业在相关药品获批后,仍需继续进行药学研发,以进一步降低风险,提高产品质量;接受基于数据和风险的灵活工艺验证方式,如应用同步验证和解耦验证策略以确保药品尽早上市,同时完成后续的验证工作;创新工具的使用需要工业界和监管机构之间进行密切的沟通交流,以尽快达成共识,降低质量风险,加快研发和审评进度。同时更好地兼顾效率与安全、风险与获益;加速研发产品上市后,通常需要通过更多的药学变更来完善,因此,制定上市后药学变更监管策略并优化审评审批程序,包括应用ICH Q12 中提到的变更工具,有助于创新药品上市后在不断提升质量的同时,保证持续稳定的市场供应。
及时研发并向患者提供高质量药物,以满足未满足的医疗需求,是全球药品监管机构和工业界的共同目标,在不影响产品质量、安全性和有效性的前提下,加速药学研发是支持加速临床研发和产品上市的关键要素之一。本文介绍的药学加速工具与ICH 相关指南中提及的基于科学和风险的原则相契合。目前,ICH正在修订部分质量相关指南,如Q1 和Q6,将明确采纳EMA 和FDA 指南中提及的药学加速工具。自2017 年加入ICH 以来,我国在推进实施ICH 指南方面取得了显著进展。希望本文能够为我国加速药学研发相关技术指导原则的研究制定及后续实施提供参考。同时,对于获得加速通道认定的产品,期待出台相应的鼓励政策,加大药品审评部门与研发企业开展前瞻性、探索性沟通交流的力度,在风险获益平衡的基础上,允许企业采取更为灵活的药学研发策略,以加速创新药物在我国的研发上市进程,造福广大患者。

来源:中国食品药品监管杂志