
嘉峪检测网 2025-08-18 21:32
导读:近日,ICH发布了ICH Q3E《可提取物和可浸出物指南》草案及其配套文件。该文件当前已完成ICH进程的第2b步,并进入第3步。该指南草案提出了可浸出杂质评估和控制的框架和流程,适用于适用于药品、生物制品、细胞和基因治疗产品,包括药械组合产品。
近日,ICH发布了ICH Q3E《可提取物和可浸出物指南》草案及其配套文件。该文件当前已完成ICH进程的第2b步,并进入第3步。该指南草案提出了可浸出杂质评估和控制的框架和流程,适用于适用于药品、生物制品、细胞和基因治疗产品,包括药械组合产品。
文件现已完成翻译,现将中英文对照分享给大家:
GUIDELINE FOR EXTRACTABLES AND LEACHABLES
ICH Q3E《可提取物和可浸出物指南》
1.INTRODUCTION
介绍
2.SCOPE
范围
3.RISK ASSESSMENT AND CONTROL OF EXTRACTABLES AND LEACHABLES
可提取物和可浸出物的风险评估和控制
3.1General Principles
一般原则
3.2Risk Matrix as a Multifactorial Concept
风险矩阵作为多因素概念
3.3Risk Assessment
风险评估
3.4Risk Control
风险控制
3.4.1Special Considerations
特殊考虑
3.5Documentation and Compliance
文件记录和合规性
3.6Risk Review / Lifecyle Management
风险审核/生命周期管理
4.CHEMICAL TESTING AND ASSESSMENT
化学测试和评估
4.1Prior Knowledge
先验知识
4.2Component Selection
组件选择
4.3Extractable Study
可提取物研究
4.3.1Semi-Quantitative Extractables Study
半定量可提取物研究
4.3.2Quantitative Extractables Study
定量可提取物研究
4.4Leachables Study
可浸出物研究
4.5Simulated Leachable Study
模拟可浸出物研究
4.6Extractable and Leachable Correlation
可提取和可浸出相关性
5.ANALYTICAL EVALUATION THRESHOLD
分析评估阈值
5.1Analytical Uncertainty Factor
分析不确定度因子
6.SAFETY ASSESSMENT
安全评估
6.1General Principles
一般原则
6.2Leachables Classification
可浸出物分类
6.3Safety Assessment Process
安全评估流程
6.4Route Specific Considerations and Special Cases (Local Toxicity Concerns)
给药途径考虑和特殊情况(局部毒性问题)
6.4.1Ophthalmic Drug Products
眼用药品
6.4.2Intracerebral, Intrathecal, Epidural Drug Products
脑内、鞘内、硬膜外药品
6.4.3Dermal Drug Products
皮肤药品
6.4.4Sensitization Potential
致敏潜力
6.5Considerations for ICH S9 Products
ICH S9产品的考虑
6.6Content of Safety Assessment
安全评估内容
7.GLOSSARY
术语
8.REFERENCES
参考文献
APPENDIX 1: TYPICAL WORKFLOWS FOR E&L RISK ASSESSMENT AND RISK CONTROL
附录1:可提取物和可浸出物风险评估和风险控制的典型工作流程
APPENDIX 2: TYPES OF STUDIES
附录2:研究类型
APPENDIX 3 AET CALCULATIONS
附录3 AET 计算
APPENDIX 4: POTENCY CLASSES FOR LEACHABLES
附录4:可浸出物的效力等级
APPENDIX 5: METHODS FOR ESTABLISHING EXPOSURE LIMITS
附录5:设定暴露限值的方法
APPENDIX 6: MONOGRAPHS FOR CLASS 1 LEACHABLES
附录6:第1类可浸出物专论
1.INTRODUCTION
介绍
Leachables are chemical entities that migrate from manufacturing components/systems, packaging or delivery device components into a drug product under the established manufacturing and labelled storage conditions. Extractables are chemical entities that are intentionally extracted from manufacturing components/systems, packaging or delivery device components under specified laboratory test conditions and thus are potential leachables.
浸出物是在既定的生产和标签标注的储存条件下,从生产组件 / 系统、包装或给药装置组件迁移到药品中的化学物质。可提取物是在特定的实验室测试条件下,从生产组件/系统、包装或给药装置组件中有意提取出的化学物质,因此可能成为浸出物。
This guideline presents a holistic framework and process for the assessment and control of leachable impurities to further expand the existing ICH guidelines on impurities, including impurities in new drug substances (ICH Q3A) and new drug products (ICH Q3B), residual solvents (ICH Q3C), and elemental impurities (ICH Q3D), as well as DNA reactive (mutagenic) impurities (ICH M7). The framework of this guideline follows the principles of risk management as described in ICH Q9. While the guideline includes materials characterization and process understanding, its primary purpose is to protect patient safety and product quality through assessment and control of leachables in the drug product. Due to rapid advances in materials engineering, device innovations, new manufacturing paradigms and novel therapeutic modalities, the aim is to provide principles and concepts that are forward looking within the scientific and regulatory landscape.
本指南提出了一个全面的框架和流程,用于评估和控制浸出性杂质,以进一步扩展现有的国际人用药品注册技术协调会(ICH)杂质指南,包括新原料药中的杂质(ICH Q3A)、新制剂中的杂质(ICH Q3B)、残留溶剂(ICH Q3C)、元素杂质(ICH Q3D)以及 DNA 反应性(致突变)杂质(ICH M7)。本指南的框架遵循 ICH Q9 中描述的风险管理原则。尽管本指南包含材料表征和工艺理解的内容,但其主要目的是通过评估和控制药品中的浸出物,保障患者安全和产品质量。由于材料工程、装置创新、新生产模式和新型治疗方式的快速发展,本指南旨在提供具有前瞻性的原则和概念,以适应科学和监管领域的发展。
2.SCOPE
适用范围
The guideline applies to the risk assessment and control of leachables in new drug products, including cell and gene therapy products. Drug-device combination products that require marketing authorizations and meet the definition of pharmaceutical or biological products are also in scope.
本指南适用于新制剂(包括细胞和基因治疗产品)中浸出物的风险评估和控制。需要上市许可且符合药品或生物制品定义的药械组合产品也在本指南的适用范围内。
Organic leachables are the primary focus of this guideline. Though recommended methodologies for elemental analysis are within the scope of this guideline, the safety assessment of elemental leachables are addressed by ICH Q3D and thus out of scope for this guideline.
有机浸出物是本指南的主要关注对象。尽管元素分析的推荐方法属于本指南的范围,但元素浸出物的安全性评估由 ICH Q3D 规范,因此不在本指南的范围内。
The guideline also applies to approved products for any changes that are likely to impact the leachable profile or patient exposure such as those relating to formulation, manufacturing, dosing, and/or container closure system (i.e., life cycle management). This guideline is not intended to apply to extrinsic, extraneous or foreign substances resulting from product contamination or adulteration.
对于已获批产品,任何可能影响浸出物特征或患者暴露量的变更(如与配方、生产、给药和 / 或容器密封系统相关的变更),本指南同样适用(即生命周期管理)。本指南不适用于由产品污染或掺假导致的外来、无关或异物。
This guideline is not intended for herbal medicinal products and crude non-processed products of animal or plant origin. For these products in liquid dosage forms, regional expectations may apply.
本指南不适用于草药制剂以及未经加工的动植物来源粗制品。对于这些液体剂型产品,可能需遵循地区性要求。
This guideline is not intended for products used during clinical research stages of development. However, in cases of high risk to the patient, principles of this guideline may be applicable to support clinical studies.
本指南不适用于临床研究阶段使用的产品。但在患者面临高风险的情况下,本指南的原则可适用于支持临床研究。
Generally, radiopharmaceuticals are not considered in scope, unless there is a specific cause for concern.
一般而言,放射性药品不在本指南的适用范围内,除非存在特定的担忧因素。
The guideline does not apply to systems used in the manufacture or storage of excipients. Refer to Section 3.4.1 for special considerations regarding packaging components for liquid or semiliquid active pharmaceutical ingredients (APIs).
本指南不适用于辅料生产或储存中使用的系统。有关液体或半液体原料药包装组件的特殊考虑,请参见第3.4.1节。
3.RISK ASSESSMENT AND CONTROL OF EXTRACTABLES AND LEACHABLES
可提取物和浸出物的风险评估与控制
3.1 General Principles
3.1 一般原则
The purpose of the guideline is to provide a holistic framework whereby leachables-associated risk can be identified, assessed, and controlled to protect the safety, efficacy, and quality attributes of the finished drug product. Figure 1 is intended to inform product development considerations leading up to product registration as well as continuous quality management process throughout lifecycle management.
本指南的目的是提供一个全面的框架,通过该框架可以识别、评估和控制与浸出物相关的风险,以保障成品药的安全性、有效性和质量属性。图 1 旨在为产品注册前的开发考量以及整个生命周期管理中的持续质量管理流程提供指导。
Figure 1: Overview of the Risk Management Process
(E&L = Extractables and Leachables)
图 1:风险管理流程概述
(E&L = 可提取物与可浸出物)
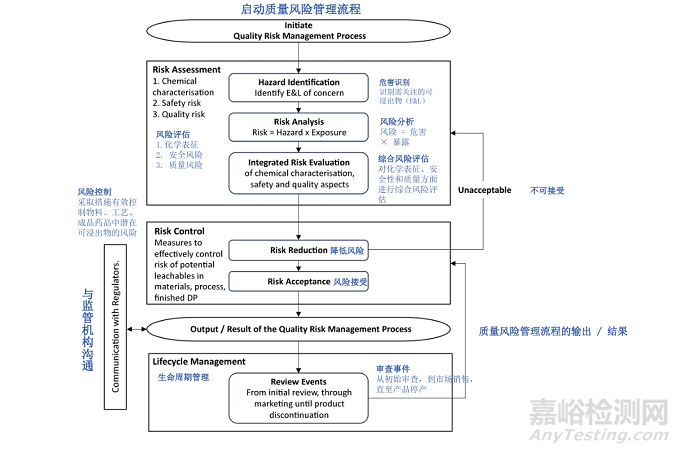
The quality risk management process for E&L warrants a holistic strategy, leveraging prior knowledge and a thorough understanding of the desirable and critical attributes for the manufacturing/packaging components and drug product, as well as the manufacturing and storage conditions. Close collaboration between the analytical chemist(s) and safety expert(s) is essential for knowledge sharing and development of the E&L quality risk management process. A Quality Risk Management Process should be initiated with every product, each with its own Risk Assessment, Risk Control and Lifecycle Management process.
可提取物和浸出物(E&L)的质量风险管理过程需要一种全面的策略,该策略应利用已有知识,并充分理解生产 / 包装组件和药品的理想属性及关键属性,以及生产和储存条件。分析化学家与安全专家之间的密切合作对于知识共享和 E&L 质量风险管理流程的制定至关重要。每个产品都应启动质量风险管理流程,且每个产品都有其自身的风险评估、风险控制和生命周期管理过程。
3.2 Risk Matrix as a Multifactorial Concept
3.2 作为多因素概念的风险矩阵
For the overall risk assessment and control of leachables, it is important to consider the multidimensional nature of risk, entailing both pharmaceutical quality and safety aspects. With respect to pharmaceutical quality, important dimensions include:
对于浸出物的整体风险评估和控制,必须考虑风险的多维度性质,这既涉及药品质量方面,也涉及安全方面。就药品质量而言,重要的维度包括
The potential for interaction between manufacturing equipment or packaging component and the formulation,
生产设备或包装组件与制剂之间发生相互作用的可能性;
The chemical and physical properties of the equipment or component that likely contribute to leachables, and pre-treatment of components prior to use,
可能导致浸出物产生的设备或组件的化学和物理性质,以及组件使用前的预处理情况;
The manufacturing and storage conditions, including but not limited to, surface area to solution volume ratio, temperature, duration of contact, proximity of the downstream removal steps and their capacity to deplete potential leachables.
生产和储存条件,包括但不限于表面积与溶液体积比、温度、接触时间、下游去除步骤的接近程度及其清除潜在浸出物的能力;
The leaching propensity of the formulation, including but not limited to API, pH, organic co-solvents and surfactant/chelating agents.
制剂的浸出倾向,包括但不限于原料药(API)、pH 值、有机助溶剂以及表面活性剂 / 螯合剂。
Safety assessment dimensions relate to the potential harms posed by leachables, inclusive of exposure-related factors such as the risk impact of the route(s) of administration, pertinent patient population(s), maximal dosing, dosing frequency and/or intervals, and maximum potential treatment duration in a lifetime.
安全评估维度涉及浸出物可能造成的潜在危害,包括与暴露相关的因素,如给药途径的风险影响、相关患者人群、最大剂量、给药频率和 / 或间隔,以及一生中潜在的最长治疗持续时间。
The relative risks associated with various dimensions (not all inclusive) are shown in Figure 2. The overall risk of a drug product is determined by taking all those dimensions into consideration.
图 2 展示了与各个维度(并非全部)相关的相对风险。药品的整体风险需综合考虑所有这些维度后确定。
Figure 2: Overview on Aspects to Consider for Risk Matrix
CSF = Cerebrospinal fluid; DP = Drug product; IM = Intramuscular; IV = Intravenous; SC = Subcutaneous
图 2:风险矩阵需考量方面概述
(CSF = 脑脊液;DP = 药品;IM = 肌内(注射);IV = 静脉(注射);SC = 皮下(注射) )
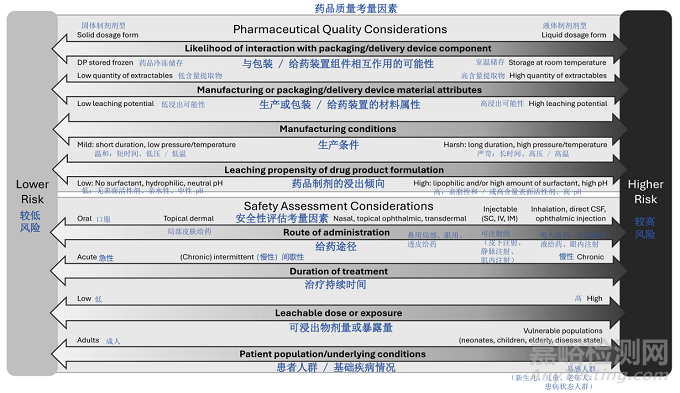
Depending on the anticipated risk and leveraging prior knowledge, various approaches can be adopted ranging from compliance with relevant food-contact safety or pharmacopeial standards/regulations to more extensive E&L characterization and safety risk assessment (See Appendix 1). For oral drug products, compliance with relevant regional food-contact safety regulations may be sufficient to support the safety and quality of polymeric manufacturing equipment/systems and container closure systems if adequately justified (e.g., proposed use is consistent with regional regulations for food contact use, the leaching propensity of the drug product is similar or less than those listed in a referenced regional regulation, and all specified testing results meet acceptance criteria). For all other drug products, or for oral products that do not comply with the regulations for food contact in terms of composition, specification, and in-use limitations, extractable/leachable assessments are typically warranted.
根据预期风险并利用已有知识,可采用多种方法,从遵守相关食品接触安全标准或药典标准 / 法规,到更广泛的可提取物和浸出物(E&L)表征及安全风险评估(见附录 1)。对于口服药品,若有充分的正当理由(例如,拟用用途符合地区食品接触用途法规,药品的浸出倾向与参考地区法规中所列的相似或更低,且所有规定的测试结果均满足验收标准),遵守相关地区的食品接触安全法规可能足以支持 polymeric 生产设备 / 系统和容器密封系统的安全性与质量。对于所有其他药品,或在成分、规格和使用限制方面不符合食品接触法规的口服药品,通常需要进行可提取物 / 浸出物评估。
The risk matrix and factors described above highlight the complexity of the risks associated with a leachables assessment. Understanding the respective risk level of the corresponding factors is part of the risk assessment process and may inform manufacturing and packaging components selection as well as the development of an overall risk assessment/control strategy.
上述风险矩阵和因素凸显了与浸出物评估相关的风险的复杂性。了解相应因素各自的风险水平是风险评估过程的一部分,可能会为生产和包装组件的选择以及整体风险评估 / 控制策略的制定提供依据。
3.3 Risk Assessment
3.3 风险评估
Based on the descriptions of the Risk Management Process (Figure 1, Section 3.1), the Multidimensional Risk Matrix (Figure 2, Section 3.2) and the Typical Workflows for E&L risk assessment and risk control (Figures 4 and 5, Appendix 1) risk assessment can be summarized in three basic steps:
根据风险管理流程(图 1,第 3.1 节)、多维度风险矩阵(图 2,第 3.2 节)以及 E&L 风险评估和风险控制的典型工作流程(图 4 和图 5,附录 1)的描述,风险评估可概括为三个基本步骤
Step 1 - Hazard Identification: Identify potential leachables that may migrate into the drug product from direct (e.g., manufacturing components/systems, container/closure systems and delivery devices components) or indirect (e.g., secondary packaging, ink or adhesives on labels particularly for semi-permeable components) contact surfaces based upon prior knowledge (experience with component, prior testing, etc.) and/or extractables and leachables testing.
步骤 1 - 危害识别基于已有知识(组件使用经验、先前测试等)和 / 或可提取物与浸出物测试,识别可能从直接接触表面(例如,生产组件 / 系统、容器 / 密封系统和给药装置组件)或间接接触表面(例如,次级包装、标签上的油墨或粘合剂,特别是对于半渗透性组件)迁移到药品中的潜在浸出物。
Step 2 - Risk Analysis: Quantitate the potential occurrence of leachables in the drug product and assess the patient exposure to leachables.
步骤 2 - 风险分析量化药品中浸出物的潜在出现情况,并评估患者对浸出物的暴露量。
Step 3 – Integrated Risk Evaluation: Evaluate the potential risk to impact product quality, safety and efficacy to determine if the selected manufacturing components/systems and container/closure systems are considered qualified for the intended use.·
步骤 3 - 综合风险评价评估可能影响产品质量、安全性和有效性的潜在风险,以确定所选生产组件 / 系统和容器 / 密封系统是否被认为适合预期用途。
3.4 Risk Control
3.4 风险控制
If the comprehensive risk assessment indicates risk mitigation is needed, measures may include, but are not limited to, change of components/suppliers, pre-wash of components, pre-flushing of manufacturing equipment, and adding additional purification/isolation step(s). The adequacy of the mitigation measures ultimately implemented should be confirmed/verified via extractable and/or leachable studies.
如果综合风险评估表明需要降低风险,所采取的措施可能包括但不限于更换组件 / 供应商、组件预清洗、生产设备预冲洗以及增加额外的纯化 / 分离步骤。最终实施的缓解措施的充分性应通过可提取物和 / 或浸出物研究进行确认 / 验证。
Once the components are qualified for the intended use, a control strategy should be implemented. This comprises, but is not limited, to routine GMP practices which are imperative for component quality controls. A control strategy should be in place to:
一旦组件被确认适合预期用途,就应实施控制策略。这包括但不限于常规的 GMP 实践,这对组件质量控制至关重要。应制定控制策略以
Establish adequate acceptance quality control including acceptance criteria, analytical procedures, and sampling plan for components as appropriate.
建立充分的验收质量控制,包括适当的组件验收标准、分析程序和取样计划。
Establish appropriate quality agreement with component venders including component lifecycle quality controls regarding any composition and/or fabrication process changes that might have impact on the extractable profiles.
与组件供应商建立适当的质量协议,包括关于可能影响可提取物特征的任何成分和 / 或制造工艺变更的组件生命周期质量控制。
See Appendix 1 for typical workflows for E&L risk assessment and risk control, including component qualifications for manufacturing components/systems (Figure 4, Appendix 1) and for packaging and delivery device components (Figure 5, Appendix 1). Typically, extractable and leachable studies should be conducted for packaging and delivery device components. Under certain circumstances alternative approaches may be proposed with proper justifications.·
有关 E&L 风险评估和风险控制的典型工作流程,包括生产组件 / 系统的组件确认(附录 1,图 4)以及包装和给药装置组件的确认(附录 1,图 5),请参见附录 1。通常,应对包装和给药装置组件进行可提取物和浸出物研究。在某些情况下,可在有充分正当理由的前提下提出替代方法。
The principles and practices used for identifying risk and developing mitigation strategies to address safety concerns associated with packaging and delivery device components are also applicable to formulation contacting manufacturing equipment components made of polymeric materials. Extractables studies should therefore be designed to represent the worst-case scenario of the manufacturing conditions (e.g., smallest scale with longest contact durations, highest temperature and pressure). It is recognized that the potential for leachables in a drug product originating from the manufacturing components/systems is lower than that from the packaging and delivery components, due to relatively shorter contacting time with the formulation and larger solution volume to surface area ratio. Leachables introduced in upstream manufacturing process steps might be able to be purged through downstream steps, e.g. purification/polish, lowering the risk for leachables ending up in the final drug product. These factors should be taken into consideration for manufacturing equipment selection and qualification, as well as quality investigations.
用于识别风险和制定缓解策略以解决与包装和给药装置组件相关的安全问题的原则和实践,也适用于与制剂接触的 polymeric 材料生产设备组件。因此,可提取物研究的设计应能代表生产条件的最坏情况(例如,最小规模、最长接触时间、最高温度和压力)。众所周知,由于与制剂的接触时间相对较短且溶液体积与表面积比较大,药品中源自生产组件 / 系统的浸出物潜力低于源自包装和给药组件的浸出物潜力。在 upstream 生产工艺步骤中引入的浸出物可能会通过下游步骤(例如纯化 / 精制)被清除,从而降低浸出物最终进入成品药的风险。在选择和确认生产设备以及进行质量调查时,应考虑这些因素。
For manufacturing components/systems, the leachables risk may be considered minimal and acceptable when all extractables peaks are at or below the Analytical Evaluation Threshold (AET) applicable to the drug product and no Class 1 leachables are observed (see Section 5). The analytical procedures used in extraction studies should comply with the criteria provided in Section 4.3.
对于生产组件 / 系统,当所有可提取物峰均等于或低于适用于该药品的分析评估阈值(AET)且未观察到 1 类浸出物时(见第 5 节),浸出物风险可被认为是最小且可接受的。提取研究中使用的分析程序应符合第 4.3 节规定的标准。
In cases where manufacturing components/systems extractables are observed in concentrations above the AET, an identification of those extractables and quantification of the concentrations may be conducted to mitigate the leachables risk as long as the quantification of extractables is performed against appropriate reference standards of the same identity as the identified extractables. However, if authentic reference standards do not exist, compounds with a similar analytical response can be employed. If extractables concentrations quantified in this manner are below the relevant acceptable safety level (see Section 6), then the safety concern associated with leachables risk is considered negligible. As an alternative to qualification of extractables from manufacturing equipment at concentrations above the AET, a safety assessment of leachables may be performed.
如果观察到生产组件 / 系统的可提取物浓度高于 AET,只要根据与已识别可提取物具有相同特性的适当参考标准对可提取物进行定量,就可以对这些可提取物进行识别和浓度定量,以降低浸出物风险。然而,如果没有真实的参考标准,可以使用具有相似分析响应的化合物。如果以这种方式定量的可提取物浓度低于相关的可接受安全水平(见第 6 节),则与浸出物风险相关的安全担忧被认为是可忽略的。作为对生产设备中浓度高于 AET 的可提取物进行确认的替代方法,可以对浸出物进行安全评估。
For a packaging component/system an abbreviated data package may be considered when patient safety risk can be adequately mitigated by prior knowledge, (e.g. established extractable/leachable correlation, similar drug product with similar leaching propensity to approved drug product formulation), or no/few extractables detected above the AET and below their applicable safety threshold (such as Class 3 leachables; See Section 6). Table A.1.2 (Appendix 1) provides examples where the overall risk is considered low, in relation to Figure 2 (Section 3.2), and an abbreviated data package may be warranted with adequate justification. When an abbreviated data package is proposed, communications with relevant regional Regulatory Agency/Health Authority is recommended to align on approach.
对于包装组件 / 系统,当通过已有知识(例如,已建立的可提取物 / 浸出物相关性、与已获批药品配方具有相似浸出倾向的类似药品)可充分降低患者安全风险,或者未检测到或仅检测到少量高于 AET 但低于其适用安全阈值的可提取物(如 3 类浸出物;见第 6 节)时,可以考虑采用简化数据包。表 A.1.2(附录 1)提供了与图 2(第 3.2 节)相关的、总体风险被认为较低且有充分理由可采用简化数据包的示例。当提出采用简化数据包时,建议与相关地区的监管机构 / 卫生当局沟通,以统一方法。
If identified extractables are likely to chemically transform into compounds with a higher safety risk (i.e. through chemical degradation and/or interaction with formulation components to generate compounds with a higher safety risk), or if not all extractable peaks above the applicable AET can be adequately identified and/or quantified, a leachable study should be conducted to address these concerns and demonstrate acceptability of the components.
如果已识别的可提取物可能通过化学降解和 / 或与制剂成分相互作用转化为具有更高安全风险的化合物,或者并非所有高于适用 AET 的可提取物峰都能被充分识别和 / 或定量,则应进行浸出物研究,以解决这些问题并证明组件的可接受性。
3.4.1 Special Considerations
3.4.1 特殊考虑因素
When multiple manufacturing components, especially those constructed with the same or similar material are used, the cumulative leachables risk should be assessed.
Quality risk assessment and derived control strategies, when appropriate, should also encompass potential leachables from a container used to store a liquid or semi-solid drug substance.
当使用多个生产组件,特别是由相同或相似材料制成的组件时,应评估累积的浸出物风险。
质量风险评估及由此制定的控制策略在适当时还应包括用于储存液体或半固体原料药的容器中可能存在的浸出物。
Although minimal leaching occurs in the frozen state, the potential for leaching from storage component/system should be evaluated before freezing and after thawing.
In addition, for biological and biotechnology-derived products risk identification and mitigation may also include:
尽管在冷冻状态下浸出量极少,但应在冷冻前和解冻后评估储存组件 / 系统的浸出潜力。
此外,对于生物制品和生物技术衍生产品,风险识别和缓解可能还包括
Evaluation of the potential interactions between reactive leachables and formulation components that may lead to potentially adverse impact on product quality, safety, and/or efficacy. If impacts to critical quality attributes of the product by known reactive leachables are identified, potential mechanisms of chemical modification should be considered (such as denaturation, aggregation or degradation).
评估反应性浸出物与制剂成分之间的潜在相互作用,此类相互作用可能对产品质量、安全性和 / 或有效性产生潜在不利影响。如果发现已知的反应性浸出物对产品的关键质量属性有影响,则应考虑化学修饰的潜在机制(如变性、聚集或降解)。
For manufacturing of drug substance, leachables may be removed during the last purification step. Therefore, the quality risk assessment will typically focus on subsequent manufacturing processes.·
在原料药生产过程中,浸出物可能在最后的纯化步骤中被去除。因此,质量风险评估通常会侧重于后续的生产工艺。
3.5 Documentation and Compliance
3.5 文件记录与合规性
Registration applications should include the justification for the extractable/leachable studies conducted, the associated study reports, the safety assessment of substances above the AET and any requisite risk control strategy. Extractables and leachables studies conducted to support the acceptability of manufacturing and packaging components/systems should be included in filing submissions (as described in ICH M4Q) as applicable. Adequate leachable data should be provided to address safety and quality concerns throughout the drug product’s shelf life. It is generally acceptable to submit leachable study results aligned with available stability data, with the provision to submit additional data post-authorization, subject to prior concurrence with the relevant regional regulatory authority. The quality risk assessment as defined in Section 3.3 of this guidance should be conducted on single-use and multi-use manufacturing components/systems, primary packaging components and delivery device components. For semi-permeable packaging materials, secondary packaging should also be evaluated as applicable.
注册申请应包括所进行的可提取物 / 浸出物研究的理由、相关的研究报告、高于分析评估阈值(AET)的物质的安全性评估以及任何必要的风险控制策略。为支持生产和包装组件 / 系统的可接受性而开展的可提取物和浸出物研究,应根据适用情况纳入申报材料中(如 ICH M4Q 所述)。应提供充分的浸出物数据,以解决药品在整个保质期内的安全性和质量问题。提交与现有稳定性数据一致的浸出物研究结果通常是可接受的,前提是在获得批准后可提交补充数据,但需事先获得相关地区监管机构的同意。本指南第 3.3 节中定义的质量风险评估应针对一次性和多次使用的生产组件 / 系统、 primary 包装组件和给药装置组件进行。对于半渗透性包装材料,还应根据适用情况对次级包装进行评估。
A list of extractables and leachables studies conducted should be included along with an assessment report which will typically include analytical method and extraction condition selections along with justifications (solvents, temperature, duration, surface/volume ratio, etc.) for extractables studies and a description of the sample preparation and analytical procedures for leachables studies. In addition, the quantification procedure(s) should be described including the suitability of the procedures used for quantification (e.g., limit of detection (LOD), limit of quantification (LOQ), specificity, linearity, accuracy, and repeatability). All extractables and leachables peaks above the AET (see Section 5) should be included in the filing submission with chemical name, structure, CAS Registry Number (if available) and observed level. For leachables (or extractables when such testing is used for qualification), safety risk assessment as described in Section 6 should be included.
申报材料中应包含所进行的可提取物和浸出物研究的清单以及评估报告,评估报告通常应包括分析方法和提取条件的选择,以及可提取物研究的理由(溶剂、温度、时间、表面积 / 体积比等),并描述浸出物研究的样品制备和分析程序。此外,还应描述定量程序,包括用于定量的程序的适用性(如检测限(LOD)、定量限(LOQ)、特异性、线性、准确度和精密度)。所有高于 AET(见第 5 节)的可提取物和浸出物峰都应纳入申报材料,并注明化学名称、结构、CAS 登记号(如可用)和观测水平。对于浸出物(或用于确认的可提取物测试),应包含第 6 节所述的安全风险评估。
In addition to the quality risk assessment, a leachables to extractables correlation should be included in the registration application, as appropriate (refer to Section 4.6). Finally, the adequacy of any proposed mitigation measures (for example prewashing of the packaging and delivery components/system or pre-flushing of the manufacturing components/systems) should be demonstrated by data collected before and after implementation.
除质量风险评估外,注册申请中还应酌情包含浸出物与可提取物的相关性分析(参见第 4.6 节)。最后,任何拟议的缓解措施(例如包装和给药组件 / 系统的预清洗或生产组件 / 系统的预冲洗)的充分性应通过实施前后收集的数据来证明。
3.6 Risk Review / Lifecycle Management
3.6 风险审查 / 生命周期管理
This section describes the types of changes that might necessitate re-evaluation of the leachable profile during the lifecycle of the drug. The following is a non-exhaustive list of potential changes and an explanation of how these represent a potential to impact the patient leachable exposure. As such, these changes should be considered and justified scientifically using new studies and/or existing information sources.
本节描述了在药品生命周期中可能需要重新评估浸出物特征的变更类型。以下是一份非详尽的潜在变更清单,并解释了这些变更如何可能影响患者对浸出物的暴露量。因此,应通过新的研究和 / 或现有信息来源,对这些变更进行科学考量和论证。
New Information: If new data and/or information on a material pertinent to its suitability for use indicates a cause for concern and/or if new patient safety information for a leachable becomes available, an updated assessment may be warranted.
新信息如果关于某一材料适用性的新数据和 / 或信息表明存在担忧因素,和 / 或出现了关于某一浸出物的新的患者安全信息,则可能需要进行更新的评估。
Changes to a drug product formulation: Changes to the drug product may cause different leachables from the existing formulation contacting manufacturing components/systems and/or primary packaging and/or delivery device components. For example, changes to excipients/surfactants composition or concentrations can affect both the composition and amount of leachables.
药品配方的变更药品的变更可能导致与现有配方接触的生产组件 / 系统和 / 或 primary 包装和 / 或给药装置组件产生不同的浸出物。例如,辅料 / 表面活性剂的成分或浓度变化可能会影响浸出物的成分和量。
Changes to container closure system, delivery device, or manufacturing components/systems that contact drug substance and/or drug product: When there are known changes such as the composition, supplier, manufacturing process, geometry or pretreatment of materials contacting the drug substance (mainly for liquids and/or biologics) or drug product during the shelf-life of the drug, there is a potential for an altered leachable profile. In addition, for some products there may be a potential for non-direct packaging components to contribute potential leachables to the drug product.
容器密封系统、给药装置或与原料药和 / 或药品接触的生产组件 / 系统的变更当在药品保质期内,与原料药(主要是液体和 / 或生物制品)或药品接触的材料在成分、供应商、生产工艺、几何形状或预处理等方面发生已知变更时,浸出物特征可能会发生改变。此外,对于某些产品,非直接接触的包装组件也可能向药品中引入潜在浸出物。
Changes to a manufacturing process: Changes to process conditions may cause different leachables or different amounts of leachables from the existing formulation contact material. For example, change in solvent system, duration, temperature, pressure, pH, cleaning/sterilization process, surface area/volume ratio, pre-operation preparation (e.g., flushing), amongst others can affect both the composition and amount of leachables.
生产工艺的变更工艺条件的变更可能导致与现有配方接触材料产生不同的浸出物或不同量的浸出物。例如,溶剂系统、时间、温度、压力、pH 值、清洁 / 灭菌工艺、表面积 / 体积比、操作前准备(如冲洗)等方面的变化,都可能影响浸出物的成分和量。
Changes that might affect patient exposure: Changes such as the posology of the drug, duration of treatment, route of administration and patient population (i.e., geriatric/pediatric) have the potential to change estimates of patient exposure to previously identified leachables, which may all affect the fundamental assumptions made in the exposure assessment and toxicological risk assessment of leachables.
可能影响患者暴露量的变更药品的剂量学、治疗持续时间、给药途径和患者人群(如老年 / 儿科患者)等方面的变更,可能会改变对患者接触先前已识别浸出物的估计,这些都可能影响在浸出物的暴露评估和毒理学风险评估中所做的基本假设。
Changes in indication that might affect patient benefit:risk: e.g. oncology to rheumatological disorders.
可能影响患者获益风险比的适应症变更例如,从肿瘤学到风湿病。
4.CHEMICAL TESTING AND ASSESSMENT
化学测试与评估
4.1 Prior Knowledge
4.1 已有知识
Prior knowledge may comprise information useful to obtain before performing chemical testing, including information available from a supplier and any relevant information with regard to other drug products and processes. This information may include:
已有知识可能包括在进行化学测试前需获取的有用信息,包括可从供应商处获得的信息以及与其他药品和工艺相关的任何信息。这些信息可能包括
composition (e.g., base polymer and copolymer, any known additives such as plasticizers, processing aids, catalysts, antioxidants)
成分(如基础聚合物和共聚物、任何已知的添加剂,如增塑剂、加工助剂、催化剂、抗氧化剂)
food contact compliance
食品接触合规性
statements indicating particular (e.g., non-authorized) compounds have not been intentionally added
表明未有意添加特定(如未经授权的)化合物的声明
compendial testing
药典测试
any available extractables studies
任何可用的可提取物研究
biological reactivity testing
生物反应性测试
processing or pretreatment steps (e.g., sterilization, cleaning, flushing, siliconization, surface treatments)
加工或预处理步骤(如灭菌、清洁、冲洗、硅化、表面处理)
prior use history, including any historical use with other similar drug products, process and/or contact conditions
既往使用历史,包括与其他类似药品、工艺和 / 或接触条件的任何历史使用情况
4.2 Component Selection
4.2 组件选择
A pharmaceutical product manufacturer is responsible for establishing requirements in alignment with regulatory expectations for the manufacturing, packaging, storage, and delivery of a unique drug product safely and effectively to an intended patient population. The level of risk for a particular material or component is relevant to the potential for interaction with the dosage form. For example, components that interact with dosage forms exhibiting a greater propensity for leaching (e.g., liquids) may be considered of higher risk than components that interact with dosage forms which exhibit a minimal propensity for leaching (e.g., non-lyophilized solids). The information obtained from the supplier (e.g., extractables report, compliance with compendial requirements) may be supplemented with additional testing appropriate for conducting a risk assessment and developing extractables/leachables procedures to demonstrate acceptable component selection. See Table A.2.1 (in Appendix 2) for a summary of extractable, leachable and simulated leachable studies.
药品制造商有责任根据监管要求,制定用于安全有效地向目标患者群体生产、包装、储存和配送特定药品的要求。特定材料或组件的风险水平与其与剂型的潜在相互作用有关。例如,与具有较高浸出倾向的剂型(如液体)接触的组件,可能被认为比与具有最小浸出倾向的剂型(如非冻干固体)接触的组件风险更高。从供应商处获得的信息(如可提取物报告、符合药典要求的证明)可辅以适当的额外测试,这些测试适用于进行风险评估和制定可提取物 / 浸出物程序,以证明组件选择的可接受性。可提取物、浸出物和模拟浸出物研究的摘要参见附录 2 中的表 A.2.1。
4.3 Extractable Study
4.3 可提取物研究
An extractable study is a process by which chemical entities are extracted from a test article. An adequate extractables study incorporates solvents and extraction conditions relevant to the anticipated leaching propensity of the drug product formulation under the worst-case scenario of manufacturing or storage conditions and employs multiple complementary analytical techniques to establish a comprehensive extractables profile. Key characteristics of an adequate extraction study include:
可提取物研究是从测试样品中提取化学物质的过程。充分的可提取物研究应采用与药品配方在生产或储存条件最坏情况下的预期浸出倾向相关的溶剂和提取条件,并采用多种互补的分析技术来建立全面的可提取物特征。充分的提取研究的关键特征包括
Establishment and application of a drug product-specific AET to indicate extractable chemical entities to be identified and treated as potential leachables. Testing is performed on components or an assembled system including any processing and treatment (e.g., sterilization, molding and fabrication conditions, cleaning, siliconization) that would be representative of the final, finished component or system as intended for use
建立并应用特定于药品的 AET,以指示需要识别并视为潜在浸出物的可提取化学物质。测试在组件或组装系统上进行,包括任何能代表最终成品组件或系统预期用途的加工和处理(如灭菌、成型和制造条件、清洁、硅化)
Proper extraction media selection, including appropriate solvents of varying pH and polarity relevant to and representative of the drug product formulation (e.g. excipients, surfactants)
适当选择提取介质,包括与药品配方相关且具有代表性的不同 pH 值和极性的适当溶剂(如辅料、表面活性剂)
Represents the drug product specific worst-case scenario for leachables occurring during manufacturing or arising from packaging components/systems during shelf life (e.g., contact area, temperature, duration)
代表药品在生产过程中产生的或在保质期内源自包装组件 / 系统的浸出物的特定最坏情况(如接触面积、温度、时间)
The analytical procedures used are adequately qualified at a level commensurate with the purpose of the extraction study
所使用的分析程序在与提取研究目的相称的水平上得到充分验证
Includes appropriate analytical procedures for volatile, semi-volatile, and non-volatile organic extractables and elemental extractables
包括针对挥发性、半挥发性和非挥发性有机可提取物以及元素可提取物的适当分析程序
The extractables report describes details on analytical procedures
可提取物报告描述分析程序的详细信息
Specific targeted tests for potential Class 1 leachables (see Section 6.2 Leachables Classification) should be performed based on the understanding of the material of construction and quality; risk analysis should be performed as appropriate. Analysis of potential Class 1 leachables should follow the description of a quantitative extractables study (Section 4.3.2) or leachables study (Section 4.4).
基于对构造材料和质量的了解,应对潜在的 1 类浸出物(见第 6.2 节浸出物分类)进行特定的靶向测试;并应酌情进行风险分析。对潜在 1 类浸出物的分析应遵循定量可提取物研究(第 4.3.2 节)或浸出物研究(第 4.4 节)的描述。
4.3.1 Semi-Quantitative Extractables Study
4.3.1 半定量可提取物研究
A semi-quantitative extractables study may be appropriate in scenarios where a leachables study will subsequently be conducted to establish the acceptability of materials for intended use. The purpose of a semi-quantitative extractables study is to understand which extractables can be present as leachables in the drug product. Key characteristics of the semi-quantitative extractables study include:
在后续将通过浸出物研究确定材料是否适合预期用途的情况下,半定量可提取物研究可能是合适的。半定量可提取物研究的目的是了解哪些可提取物可能作为浸出物存在于药品中。半定量可提取物研究的关键特征包括
Analytical procedures that are qualified using several relevant standard compounds typically observed as extractables or leachables.
使用几种通常被视为可提取物或浸出物的相关标准化合物对分析程序进行验证。
Use of analytical uncertainty factor (UF; Section 5.1) in the calculation of the drug product-specific AET.
在计算特定于药品的分析评估阈值(AET)时使用分析不确定度因子(UF;第 5.1 节)。
Quantification of observed extractables against relevant standard compounds.
根据相关标准化合物对观察到的可提取物进行定量。
Semi-quantitative extractables observed above the AET can subsequently be used as targets for a quantitative extractables study or a leachables study.·
观察到的高于 AET 的半定量可提取物可随后作为定量可提取物研究或浸出物研究的目标。
4.3.2 Quantitative Extractables Study
4.3.2 定量可提取物研究
To support qualification of manufacturing components/systems and certain low-risk packaging components/systems scenarios (Refer to Appendix 1 Table A.1.1 and A.1.2, respectively) for which extractables were observed at a level above the AET during the semi-quantitative extractables study, a quantitative extractables study to quantify these specific extractables would be warranted. Key characteristics of quantitative extractables study include:
为支持生产组件 / 系统和某些低风险包装组件 / 系统的确认(分别参见附录 1 表 A.1.1 和 A.1.2),若在半定量可提取物研究中观察到可提取物水平高于 AET,则需要进行定量可提取物研究以量化这些特定可提取物。定量可提取物研究的关键特征包括
Confirmed identification of extractables above the AET.
确认识别出高于 AET 的可提取物。
Quantification of the identified extractables above the AET using standards with identical or similar analytical response.
使用具有相同或相似分析响应的标准品对已识别的高于 AET 的可提取物进行定量。
The analytical procedure used for quantifying the identified extractables above the AET should be qualified for the specific standard compound.
用于量化已识别的高于 AET 的可提取物的分析程序应针对特定标准化合物进行验证。
If the amount of an adequately identified and quantified extractable exceeds its qualification limit (e.g., applicable safety threshold or permitted daily exposure (PDE)), a leachables study is warranted to demonstrate the compound as a leachable remains below its qualification limit. In addition, a leachables study can also be used to assess the quality risk for extractables above the AET when those extractables cannot be identified with confirmed identities.·
如果充分识别和定量的可提取物量超过其确认限度(如适用的安全阈值或允许日暴露量(PDE)),则需要进行浸出物研究,以证明该化合物作为浸出物的水平仍低于其确认限度。此外,当无法确认高于 AET 的可提取物的身份时,浸出物研究也可用于评估其质量风险。
4.4 Leachables Study
4.4 浸出物研究
Leachables studies intended to support drug product registration are designed to represent the actual manufacturing conditions and intended storage conditions throughout the proposed shelf-life and in-use period. During the shelf life and in-use period, multiple time points should be evaluated to characterize trending of leachables to estimate maximal occurrence. The leachables assessment for the container closure system is performed on the actual drug product during stability storage and may include accelerated storage conditions. For a container closure system, the study should involve multiple primary drug product stability and/or development batches manufactured with the actual packaging and delivery system intended for use with the commercial product. If multiple batches are not available, alternative approaches may be proposed with justification. Use of the same lots of components used in extractables assessments potentially enables a more meaningful correlation between extractables and leachables. Analytical procedures for specific, targeted leachables should be appropriately validated to establish that they are sensitive, selective, accurate, and precise. Non-targeted screening procedures should also be used and employ appropriate analytical techniques to facilitate detection of any unanticipated degradation of leachables, leachables from secondary packaging, and/or interaction products. The non-targeted screening study should include the application of an AET (See Section 5) to indicate a level above which leachable chemical entities should be identified, quantified, and reported for toxicological assessment.
旨在支持药品注册的浸出物研究旨在模拟实际生产条件以及拟议保质期和使用期间的预期储存条件。在保质期和使用期间,应评估多个时间点以表征浸出物的趋势,从而估计其最大出现量。容器密封系统的浸出物评估在药品稳定性储存期间进行,可能包括加速储存条件。对于容器密封系统,研究应涉及多个主要药品稳定性批次和 / 或开发批次,这些批次使用与商业产品预期一致的实际包装和给药系统生产。如果无法获得多个批次,可在提供正当理由的情况下提出替代方法。使用与可提取物评估相同批次的组件可能有助于在可提取物和浸出物之间建立更有意义的相关性。针对特定目标浸出物的分析程序应进行适当验证,以确保其灵敏、特异、准确和精密。还应使用非靶向筛选程序,并采用适当的分析技术,以促进检测任何未预期的浸出物降解、来自次级包装的浸出物和 / 或相互作用产物。非靶向筛选研究应应用 AET(见第 5 节),以指示高于该水平的浸出化学物质应被识别、定量并报告以进行毒理学评估。
Reference standards, if available, are preferred as they facilitate more accurate and precise quantitation of target leachables that may be present as actual drug product leachables when they are used to produce either proper response factors or calibration curves; in which case the analytical accuracy and precision is high.
如果有参考标准品,应优先使用,因为当它们用于生成适当的响应因子或校准曲线时,有助于更准确和精密地定量可能作为药品实际浸出物存在的目标浸出物;在这种情况下,分析的准确度和精密度较高。
4.5 Simulated Leachable Study
4.5 模拟浸出物研究
Circumstances may exist when performing a drug product leachables study is not technically feasible despite thorough due diligence which may include systematic investigation of multiple diverse sample preparation techniques coupled with highly sensitive and selective analytical methods, techniques and instrumentation. Such circumstances may include challenging detection or quantification thresholds associated with large volume parenterals (LVPs), significant analytical matrix interference inherent with complex drug product formulations, or a combination of such factors. In such situations, use of a simulation study to support actual drug product leachables evaluation may be justifiable. For example, a simulation study could be performed to augment a leachables study to accomplish the objectives that cannot be obtained by leachables testing. In the case of a challenging AET (i.e., procedure LOQ > AET), the leachables study would be performed with relevant test procedure LOQ and a simulation study would be performed to fill in the gap between the LOQ and the AET. Alternatively, a simulation study could be used to replace a leachables study when, through thorough due diligence, it is established that performing the leachables study is impractical.
尽管已尽到应有的努力(可能包括系统研究多种不同的样品制备技术,结合高灵敏度和高选择性的分析方法、技术和仪器),但在某些情况下,进行药品浸出物研究在技术上仍不可行。此类情况可能包括与大容量注射剂(LVPs)相关的检测或定量阈值挑战、复杂药品配方固有的显著分析基质干扰,或此类因素的组合。在这种情况下,使用模拟研究来支持实际药品浸出物评估可能是合理的。例如,可进行模拟研究以补充浸出物研究,以实现浸出物测试无法实现的目标。在 AET 难以达到的情况下(即程序定量限(LOQ)> AET),浸出物研究将使用相关测试程序的 LOQ 进行,而模拟研究将用于填补 LOQ 与 AET 之间的差距。或者,当通过彻底的尽职调查确定进行浸出物研究不切实际时,模拟研究可用于替代浸出物研究。
It is important to recognize that, regardless of how well the simulation study is designed and executed, its outcome will likely only approximate the results of a drug product leachable study and cannot fully replicate a true leachable profile of the drug product. For example, a simulation study cannot and will not address any potential interaction between leachables and the components of the drug product formulation components.
重要的是要认识到,无论模拟研究设计和执行得多么完善,其结果可能只能近似于药品浸出物研究的结果,无法完全复制药品的真实浸出物特征。例如,模拟研究无法也不会解决浸出物与药品配方成分之间的任何潜在相互作用。
The simulation study is a surrogate study that reveals likely true leachables that would be detected if a leachables study could have been conducted. Thus, the simulated leachables detected above the simulation study’s drug product specific AET should be identified, quantified, and assessed for safety. As the goal of a simulation study is to obtain a simulated leachables profile that closely mimics the actual leachables profile generated by the drug product over its shelf-life, the simulation conditions and process used in the simulation study should closely match the drug product manufacturing/storage conditions used in a leachables study, with the intent of simulating the conditions experienced by the drug product during its manufacturing, shelf-life storage, and in-use (clinical) preparation. Furthermore, the simulation solvent should be chosen so that is has a similar propensity to leach as the drug product, and the simulated manufacturing process should be performed using worst-case conditions. Moreover, a simulation study can be accelerated versus drug product shelf storage conditions to mimic the outcome of a leachable study over the entire drug product shelf life with shorter duration.
模拟研究是一种替代研究,它揭示了如果能够进行浸出物研究可能检测到的潜在真实浸出物。因此,在模拟研究中检测到的高于特定药品 AET 的模拟浸出物应被识别、定量并进行安全性评估。由于模拟研究的目标是获得一个接近模拟药品在其保质期内产生的实际浸出物特征的模拟浸出物特征,因此模拟研究中使用的模拟条件和过程应与浸出物研究中使用的药品生产 / 储存条件密切匹配,以模拟药品在生产、保质期储存和使用(临床)制备过程中所经历的条件。此外,选择的模拟溶剂应具有与药品相似的浸出倾向,并且模拟生产过程应在最坏情况下进行。此外,与药品保质期储存条件相比,模拟研究可以加速进行,以在更短的时间内模拟整个药品保质期内的浸出物研究结果。
As the intent of the simulation study is to augment or replace a leachables study, the simulation study must meet all the quality requirements for a leachables study, including test procedure qualification. When properly justified, use of a simulation study is an alternative to the recommended practice of performing leachables studies. Thus, the intended application, justification, and qualification of a simulated leaching study for a particular drug product should be based on a scientifically sound rationale with demonstration of due diligence supported by appropriate testing and experimentation. When considering the use of a simulation study, consultation with the relevant regional Regulatory Agency prior to implementation may be warranted.
由于模拟研究的目的是补充或替代浸出物研究,因此模拟研究必须满足浸出物研究的所有质量要求,包括测试程序验证。在有充分正当理由的情况下,使用模拟研究可作为进行浸出物研究这一推荐做法的替代方案。因此,针对特定药品的模拟浸出研究的预期应用、理由和确认应基于科学合理的依据,并通过适当的测试和实验证明已尽到应有的努力。在考虑使用模拟研究时,可能需要在实施前与相关地区的监管机构进行咨询。
4.6 Extractable and Leachable Correlation
4.6 可提取物与浸出物的相关性
The main purpose for generating extractables profiles is to characterize and assist selection of components, identify potential leachables, develop methods for targeted leachables, and correlate leachables and extractables. Leachables generally represent a subset of the extractables and the concentration of each leachable is typically below that of the corresponding extractable from a well conducted extractables study.
生成可提取物特征的主要目的是表征和辅助组件选择、识别潜在浸出物、开发目标浸出物的检测方法,以及建立浸出物与可提取物之间的相关性。浸出物通常是可提取物的一个子集,且在设计良好的可提取物研究中,每种浸出物的浓度通常低于相应可提取物的浓度。
Once the E&L profiles above AET are available, it is recommended that a qualitative and quantitative correlation between the two be evaluated. A correlation between leachables and extractables may be established when actual drug product leachables can be comparatively linked qualitatively and quantitatively with extractables from corresponding extractables studies of components or systems. Correlating leachables with extractables may support a justification for the use of routine extractables testing of components as an alternative to routine leachables testing during stability studies when appropriate for high-risk drug products, change control, and ongoing quality control. Potential explanations for leachables that were not detected or detected at higher levels than suggested by the extraction study conditions could include inadequate design and/or execution of the extractables study, degradation of leachables to form new compounds, interaction products of leachables with API and/or excipients, chemicals migrated from packaging, and/or new leachables resulting from materials change due to aging (e.g., exposure to UV light, heat, oxygen) during shelf-life storage. Though the E&L correlation is valuable and informative for the quality risk assessment and may be leveraged for component selection and life-cycle management decisions, it is the leachables profile that ultimately drives patient safety risk evaluations and component acceptability.
一旦获得高于 AET 的可提取物和浸出物(E&L)特征,建议评估两者之间的定性和定量相关性。当药品实际浸出物可与组件或系统的相应可提取物研究中的可提取物进行定性和定量对比关联时,可建立浸出物与可提取物之间的相关性。在高风险药品的稳定性研究、变更控制和持续质量控制中,将浸出物与可提取物相关联可能为使用组件的常规可提取物测试替代常规浸出物测试提供依据。对于未检测到浸出物或检测到的浸出物水平高于提取研究条件所显示的水平,可能的解释包括可提取物研究设计和 / 或执行不足、浸出物降解形成新化合物、浸出物与原料药和 / 或辅料的相互作用产物、从包装迁移的化学物质,以及 / 或在保质期储存期间因材料老化(如暴露于紫外线、热、氧气)而产生的新浸出物。尽管 E&L 相关性对于质量风险评估具有价值和参考意义,并可用于组件选择和生命周期管理决策,但最终决定患者安全风险评估和组件可接受性的是浸出物特征。
Any changes occurring during the product life-cycle significantly altering the extractable/leachable profiles should prompt re-evaluation of the extractable/leachable profiles and their correlation. If a specific leachable is observed in the drug product during stability studies at a level significantly greater than anticipated from the calculated potential maximum level of the leachable as established with the extraction study conducted on the same component/system lots as were used for the drug product stability batches, it can indicate that the extraction study was incomplete and it may not be possible to establish a meaningful leachables to extractables correlation for that particular leachable.
在产品生命周期中发生的任何显著改变可提取物 / 浸出物特征的变更,都应促使对可提取物 / 浸出物特征及其相关性进行重新评估。如果在稳定性研究中,药品中观察到的特定浸出物水平显著高于根据对与药品稳定性批次所用相同组件 / 系统批次进行的提取研究确定的浸出物潜在最大计算水平,则可能表明提取研究不完整,且可能无法为该特定浸出物建立有意义的浸出物与可提取物相关性。
5.ANALYTICAL EVALUATION THRESHOLD
分析评估阈值
The AET is not a control threshold, but rather a threshold corresponding to a concentration above which extractables or leachables should be identified, quantitated, and reported for safety assessment, forming the foundation of the overall E&L risk assessment and control strategy. The ICH guidelines on impurities in new drug substances (ICH Q3A) and impurities in new drug products (ICH Q3B), describe a series of predetermined thresholds based upon maximum daily dosing that are intended to provide adequate control over critical quality attributes that may impact the safety and efficacy of the drug product over the course of the product shelf-life. In contrast, this guideline recommends incorporation of a Safety Concern Threshold (SCT; see Section 6 Safety Assessment) to first establish a study-specific AET.
分析评估阈值(AET)不是控制阈值,而是一个浓度阈值,高于该阈值的可提取物或浸出物应被识别、定量并报告以进行安全评估,它构成了整体可提取物和浸出物(E&L)风险评估和控制策略的基础。国际人用药品注册技术协调会(ICH)关于新原料药中的杂质(ICH Q3A)和新制剂中的杂质(ICH Q3B)的指南,描述了一系列基于最大日剂量的预定阈值,旨在充分控制可能影响药品在保质期内安全性和有效性的关键质量属性。相比之下,本指南建议纳入安全关注阈值(SCT;见第 6 节安全评估),以首先建立特定于研究的 AET。
An extraction study should include the establishment and application of an AET to indicate extractable chemical entities to be detected, identified and reported as potential leachables for the drug product. For a leachable study, the AET is established at a concentration above which compounds should be identified and quantitated to enable appropriate safety assessment. For Class 1 leachables (See Appendix 4, Table A.4.1), the compound-specific safety limit, instead of a product-specific SCT, should be used for quantification.
提取研究应包括分析评估阈值(AET)的建立和应用,以指示需要检测、识别并报告为药品潜在浸出物的可提取化学物质。对于浸出物研究,AET 设定为一个浓度阈值,高于该浓度的化合物应被识别和定量,以便进行适当的安全评估。对于 1 类浸出物(参见附录 4 表 A.4.1),应使用化合物特异性安全限值而非产品特异性安全关注阈值(SCT)进行定量。
Derivation of the study-specific AET depends on dosing considerations (e.g., maximum dose level, frequency of dosing, and duration of treatment). The AET may be expressed using various units of measure depending on the type of study (extractable vs leachable) and what is being evaluated. For example, weight of extractable per weight of component material (e.g., µg/g) or weight of extractable per extraction solution volume (e.g., µg/mL) are commonly used units for extractables in solutions. For leachables studies, weight of leachables per packaging or delivery component/system (e.g., µg/component, µg/mL, µg/g, ppm) may be used to represent the leachables AET based on the entire container closure system or set of manufacturing components. Regardless of the units used to express the AET, they will all equate to an equivalent potential patient dose for a given study. Example AET calculations are presented in Appendix 3.
特定于研究的 AET 的推导取决于给药因素(如最大剂量水平、给药频率和治疗持续时间)。根据研究类型(可提取物研究 vs 浸出物研究)和评估对象的不同,AET 可采用多种计量单位。例如,溶液中可提取物常用的单位有每重量组件材料中的可提取物重量(如 µg/g)或每体积提取溶液中的可提取物重量(如 µg/mL)。对于浸出物研究,可基于整个容器密封系统或整套生产组件,采用每包装或给药组件 / 系统中的浸出物重量(如 µg / 组件、µg/mL、µg/g、ppm)来表示浸出物 AET。无论用于表示 AET 的单位如何,对于特定研究,它们都等同于潜在的患者等效剂量。附录 3 提供了 AET 计算示例。
5.1 Analytical Uncertainty Factor
5.1 分析不确定度因子
When an AET is used in semi-quantitative analytical methods, an appropriate uncertainty factor should be applied to account for potential underestimation of analyte concentrations due to differences in response factors between analytes and the reference standard.
当在半定量分析方法中使用 AET 时,应应用适当的不确定度因子,以考虑由于分析物与参考标准品之间的响应因子差异而可能导致的分析物浓度低估。
The determination of the appropriate magnitude for the analytical uncertainty factor(s) in a given extractable/leachable study depends on the prior knowledge and understanding of the materials of construction, the possible chemical structure of the potential extractables/leachables, the availability of the reference standards covering the range of response factors, and the limitations of the analytical methods.
在特定的可提取物 / 浸出物研究中,分析不确定度因子的适当大小取决于对构造材料的已有知识和理解、潜在可提取物 / 浸出物的可能化学结构、涵盖响应因子范围的参考标准品的可获得性,以及分析方法的局限性。
Under certain circumstances an acceptable approach is to multiply an uncertainty factor (UF) of no greater than 0.5. Alternatively, an uncertainty factor can be derived from statistical analysis of appropriately constituted response factor database of relevant reference compounds. Justification of UF applied should be included in the extractable/leachable study report.
在某些情况下,可接受的做法是采用不大于 0.5 的不确定度因子(UF)进行乘法运算。或者,可通过对相关参考化合物的适当构成响应因子数据库进行统计分析来推导不确定度因子。可提取物 / 浸出物研究报告中应包含所应用的 UF 的论证依据。
6.SAFETY ASSESSMENT
安全评估
6.1 General Principles
6.1 一般原则
A risk-based scientific evaluation is needed to provide confidence that any potential leachables in the drug product are at levels where they pose negligible risk to the patient. Within this overall risk-based evaluation, the focus of the safety assessment is the toxicological evaluation of leachables in the drug product exceeding a predefined SCT for that drug product. Within this context, the SCT is considered the threshold below which a leachable would have an exposure so low as to present negligible mutagenic and non-mutagenic toxicity concerns. The outcome of the safety assessment can be used to determine if levels of Class 1 leachables from a material are considered acceptable and may be used to set specifications for leachables in the drug product if needed.
需要进行基于风险的科学评估,以确保药品中任何潜在浸出物的水平对患者构成的风险可忽略不计。在这种基于风险的整体评估中,安全评估的重点是对药品中超过该药品预定义 SCT 的浸出物进行毒理学评估。在此背景下,SCT 被视为一个阈值,低于该阈值时,浸出物的暴露量极低,以至于其致突变性和非致突变性毒性担忧可忽略不计。安全评估的结果可用于确定某一材料中 1 类浸出物的水平是否可接受,并可在需要时用于设定药品中浸出物的规格。
Since the SCT is defined to be protective of both mutagenic and non-mutagenic effects, it must consider both mutagenicity concerns and concerns related to alternative toxicity endpoints and is based on whichever is more limiting with respect to exposure. As such, in addition to amount of exposure, the SCT dependent on both route and duration of exposure. For mutagenicity concerns, the Threshold of Toxicological Concern (TTC) as described in ICH M7 is considered applicable. For non-mutagenic toxicity endpoints, a Qualification Threshold (QT) is used in this guideline and may be considered as a dose at which potential non-mutagenic toxic effects are negligible. Subsequently, the SCT is the lowest value of either the TTC or QT for a specific drug product, considering route and potential duration of exposure. Oral and parenteral QT values have been derived by review of approximately 330 potential leachable permitted daily exposures (PDEs). An overview of these systemic safety thresholds (expressed in µg/day) for oral, parenteral, dermal/transdermal and inhalation routes of exposure, are provided in Table 1. In addition, local toxicity thresholds for leachable concentrations in drug products for topical ophthalmic, subcutaneous/intradermal, dermal/transdermal and inhalation routes of exposure are presented. For other routes of administration, the concepts described in this guideline may be used to determine acceptable exposure levels.
由于 SCT 被定义为可同时防范致突变和非致突变效应,因此它必须同时考虑致突变性担忧和与其他毒性终点相关的担忧,并基于在暴露方面更具限制性的因素。因此,除暴露量外,SCT 还取决于暴露途径和持续时间。对于致突变性担忧,ICH M7 中描述的毒理学关注阈值(TTC)被认为是适用的。对于非致突变性毒性终点,本指南使用确认阈值(QT),该阈值可被视为潜在非致突变性毒性效应可忽略不计的剂量。因此,对于特定药品,SCT 是考虑暴露途径和潜在暴露持续时间后,TTC 或 QT 中的较低值。通过对约 330 种潜在浸出物的允许日暴露量(PDE)进行审查,得出了口服和胃肠外给药途径的 QT 值。表 1 提供了口服、胃肠外、皮肤 / 经皮和吸入给药途径的全身安全阈值(以 µg / 天表示)概述。此外,还列出了局部眼科、皮下 / 皮内、皮肤 / 经皮和吸入给药途径的药品中浸出物浓度的局部毒性阈值。对于其他给药途径,可使用本指南中描述的概念来确定可接受的暴露水平。
Table 1: Systemic and Local Toxicity Thresholds
表 1:全身毒性与局部毒性阈值
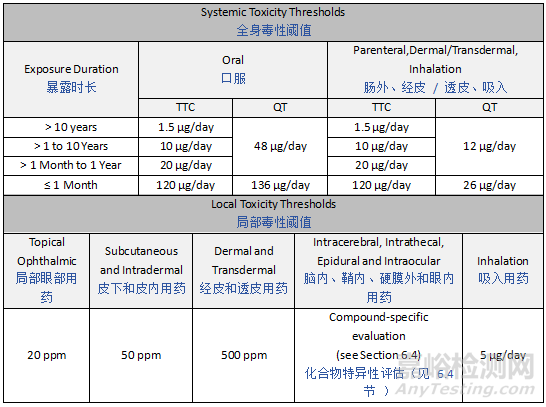
QT values for inhalation and dermal/transdermal routes have been established based upon parenteral QT in lieu of available PDE values.
吸入和皮肤/经皮给药途径的确认阈值(QT)是基于胃肠外给药的QT确立的,以替代现有的允许日暴露量(PDE)数值。
6.2 Leachables Classification
6.2 浸出物分类
Potential leachables from various materials encompass a large variety of chemicals, and thus toxicological characteristics. To provide a pragmatic, risk-based approach to leachables safety assessment, certain compounds need to be controlled at levels that are lower than the established qualification threshold due to their potential for highly potent toxicity. Such chemicals are categorized as Class 1 leachables in the current guideline. For mutagenic carcinogens, the Cohort of Concern as defined in ICH M7 and ICH M7 Class 1 impurities with an AI below 1.5 µg/day are considered Class 1 leachables. Similarly, there are some compounds, such as bisphenol A (BPA) or benzo(a)pyrene, that may have potent non-mutagenic toxicity concerns that may theoretically be associated with a greater than negligible patient safety risk at or below the drug product QT value. For such Class 1 leachables, it is considered most practical to avoid the use of materials which may leach such compounds (see Section 5). However, if the use of such materials or components is considered unavoidable, a compound-specific safety limit for these substances should be used.
来自各种材料的潜在浸出物包含多种化学物质,因此具有不同的毒理学特征。为了对浸出物安全评估采用务实的、基于风险的方法,某些化合物由于具有强效毒性潜力,需要控制在低于既定确认阈值的水平。此类化学物质在本指南中被归类为1类浸出物。对于致突变性致癌物,ICH M7中定义的“关注队列”(Cohort of Concern)以及AI低于1.5 µg/天的ICH M7 1类杂质均被视为1类浸出物。类似地,某些化合物(如双酚A(BPA)或苯并(a)芘)可能具有强效非致突变性毒性,理论上在药品QT值或以下水平时,可能对患者安全构成不可忽略的风险。对于此类1类浸出物,最切实的做法是避免使用可能浸出这些化合物的材料(见第5节)。然而,如果认为无法避免使用此类材料或组件,则应采用这些物质的化合物特异性安全限值。
Class 3 leachables are compounds established to have relatively low potency for systemic toxicity with derived chronic parenteral PDEs in excess of the levels at which leachables are typically observed (i.e., PDE ≥ 1 mg/day using the methodology described in Appendix 5). Class 3 leachables would not require further safety qualification if observed at daily exposure levels < 1 mg/day. In between these two classes are compounds with a toxicity potential that may be relevant at levels commonly encountered for leachables (Class 2 leachables). Appendix 4 provides an overview of these three leachable classes.
3类浸出物是被证实具有相对较低全身毒性潜力的化合物,其推导的慢性胃肠外给药允许日暴露量(PDE)超过浸出物通常被观察到的水平(即使用附录5中描述的方法得出的PDE ≥ 1 mg/天)。如果3类浸出物的日暴露水平< 1 mg/天,则无需进一步的安全确认。介于这两类之间的是在浸出物常见水平下可能具有相关毒性潜力的化合物(2类浸出物)。附录4概述了这三类浸出物。
6.3 Safety Assessment Process
6.3 安全评估流程
Organic leachables exceeding the AET should be identified, quantified, and reported for safety risk assessment. Acceptability of partial or incomplete elucidation of the compound structure should be justified from an analytical perspective. If toxicologically justified, partial elucidation providing tentative structures may inform a safety assessment in certain cases. The general process for safety assessment of leachables is presented in a flowchart (Figure 3) and includes an assessment of both mutagenicity and general toxicity concerns.
超过分析评估阈值(AET)的有机浸出物应被识别、定量并报告,以进行安全风险评估。化合物结构的部分或不完全解析的可接受性应从分析角度进行论证。在某些情况下,如果有毒理学依据,提供暂定结构的部分解析可能有助于安全评估。浸出物安全评估的一般流程如图3所示,包括对致突变性和一般毒性的评估。
Figure 3: Safety Assessment Process for Leachables Using Safety Evaluation Thresholds
图 3:利用安全性评估阈值开展可浸出物安全性评估流程
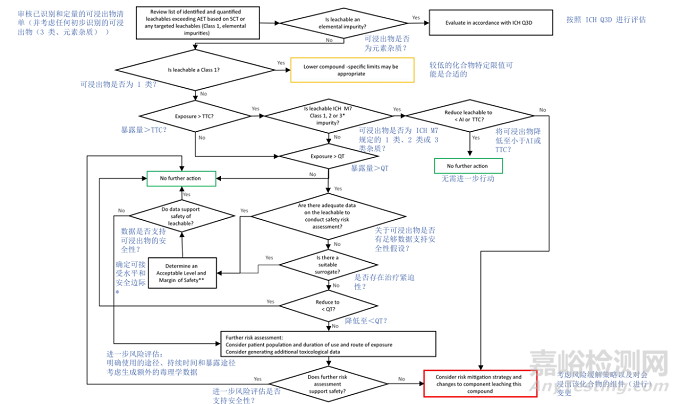
As described in ICH M7.
** If daily exposure to leachable is >1 mg/day, genotoxicity studies should be considered, as recommended in ICH Q3A and ICH Q3B (e.g., bacterial mutagenicity study and in vitro chromosomal aberration assay).
如 ICH M7 中所述。
** 若浸出物的日暴露量 > 1 mg / 天,应考虑进行遗传毒性研究,如 ICH Q3A 和 ICH Q3B 中建议的(例如,细菌致突变性研究和体外染色体畸变试验)。
Potential Class 1 leachables should ideally be identified and avoided during materials and component selection. However, if such compounds cannot be avoided, lower compound-specific thresholds and specifications to adequately control their presence as leachables should be implemented as an initial step in the process. Subsequently, all leachables above the TTC applicable to the drug product should be evaluated for mutagenic potential according to ICH M7. Leachables considered potentially mutagenic should be appropriately controlled within TTC limits unless de-risked by appropriate mutagenicity studies.
在材料和组件选择过程中,理想情况下应识别并避免潜在的 1 类浸出物。然而,若无法避免此类化合物,作为流程的初始步骤,应实施更低的化合物特异性阈值和规格,以充分控制其作为浸出物的存在。随后,所有高于药品适用毒理学关注阈值(TTC)的浸出物应根据 ICH M7 评估其致突变潜力。被认为具有潜在致突变性的浸出物应在 TTC 限值内进行适当控制,除非通过适当的致突变性研究降低了风险。
In addition to the mutagenicity assessment, all leachables above the applicable QT for the drug product should also be evaluated for general toxicity concerns. If adequate data are available to support the safety of the leachable at the maximal potential patient exposure, then no further toxicological assessment is needed (See Appendix 5 for further information). Conversely, if data do not sufficiently support the safety of the leachable, further action is needed to reduce the potential exposure to a known acceptable level (material replacement, etc.), generation of additional toxicological data to qualify the observed level, or a risk/benefit assessment providing justification of exposure at the observed level.
除致突变性评估外,所有高于药品适用确认阈值(QT)的浸出物还应评估其一般毒性风险。若有充分数据支持浸出物在患者潜在最大暴露量下的安全性,则无需进一步的毒理学评估(详见附录 5)。相反,若数据不足以支持浸出物的安全性,则需要采取进一步措施将潜在暴露量降低至已知可接受水平(如更换材料等)、生成额外的毒理学数据以确认观察到的水平,或进行风险 / 获益评估以证明观察到的暴露水平的合理性。
It should be noted that for leachables where adequate data to inform on the safety of the compound are not available, a read across approach using a highly similar compound(s) with toxicological data is encouraged. If suitable surrogate(s) can be identified that have sufficient data to support the safety of the observed leachable at the level observed, further safety risk assessment and/or studies can be avoided.
应注意,对于缺乏充分数据说明其安全性的浸出物,鼓励采用“交叉参照” 法,即使用具有毒理学数据的高度相似化合物进行评估。若能确定合适的替代物,且其数据足以支持观察到的浸出物在该水平下的安全性,则可避免进一步的安全风险评估和 / 或研究。
If the generation of novel toxicological data is considered necessary to support the safety of exposure to a leachable, New Approach Methodologies (NAMs) including in silico and in vitro models may be considered if appropriately justified. Otherwise, a toxicological qualification study(ies) as described in ICH Q3A and Q3B should be considered in order support safety assessment of the compound(s).
若认为需要生成新的毒理学数据以支持浸出物暴露的安全性,在有充分理由的情况下,可考虑采用新方法学(NAMs),包括计算机模拟和体外模型。否则,应考虑按照 ICH Q3A 和 Q3B 中描述的毒理学确认研究,以支持对化合物的安全评估。
6.4 Route Specific Considerations and Special Cases (Local Toxicity Concerns)
6.4 特定给药途径的考虑因素和特殊情况(局部毒性关注)
Safety risk assessments for potential systemic toxicity are typically sufficient to support the safety of exposure to leachables. However, there are certain scenarios where potential local toxicity effects may be pertinent due to the potential for damage to vulnerable tissues related to the local concentration of a compound (e.g., pulmonary drug products, ophthalmic drug products, and intracerebral/intrathecal/epidural drug products). When relevant, the toxicological risk assessment should address the potential impact of a leachable on local tissue toxicity as well as factors that may potentially reduce such concerns (e.g., formulation and excipients, contact duration, recovery of tissue damage). Additionally, when potential local toxicity needs to be considered, the SCT used should be the lowest (on a daily exposure basis) of the mutagenic (i.e., TTC), non-mutagenic (i.e., QT), and local toxicity thresholds (pertinent concentration converted to a maximum daily exposure level).
对潜在全身毒性的安全风险评估通常足以支持浸出物暴露的安全性。然而,在某些情况下,由于化合物的局部浓度可能对脆弱组织造成损害,潜在的局部毒性效应可能更为相关(例如,肺部用药品、眼科用药品以及脑内 / 鞘内 / 硬膜外用药品)。在相关情况下,毒理学风险评估应说明浸出物对局部组织毒性的潜在影响,以及可能降低此类担忧的因素(如配方和辅料、接触时间、组织损伤的恢复)。此外,当需要考虑潜在局部毒性时,所使用的安全关注阈值(SCT)应为(基于日暴露量)致突变性阈值(即 TTC)、非致突变性阈值(即 QT)和局部毒性阈值(相关浓度转换为最大日暴露水平)中的最低值。
6.4.1 Ophthalmic Drug Products
6.4.1 眼科用药品
Ophthalmic products are often administered topically, while some products are injected directly into ocular tissues. There is a paucity of data to characterize the potential local toxicity of leachables when in contact with ocular tissues. Based on historical precedence, in the absence of a relevant database, a compound-specific risk assessment should be completed for topically administered products to justify the safety of a leachable when it exceeds a concentration of 20 ppm in the final to-be-marketed topical ophthalmic products. This concentration limit is not considered applicable to irrigation fluids that are in transient contact with ocular tissues. For products injected into ocular tissues no threshold is given. A qualitative safety assessment of any leachables present should be provided, since such leachables may be of relevance even when present at a concentration below 20 ppm.
眼科用药品通常为局部给药,部分产品则直接注射到眼部组织中。关于浸出物与眼部组织接触时的潜在局部毒性,相关数据较为匮乏。根据历史先例,在缺乏相关数据库的情况下,对于局部给药产品,当浸出物在最终上市的局部眼科用药品中的浓度超过 20 ppm 时,应完成化合物特异性风险评估,以证明其安全性。该浓度限值不适用于与眼部组织短暂接触的冲洗液。对于注射到眼部组织的产品,未设定阈值。应对存在的任何浸出物进行定性安全评估,因为即使浓度低于 20 ppm,此类浸出物也可能具有相关性。
6.4.2 Intracerebral, Intrathecal, Epidural Drug Products
6.4.2 脑内、鞘内、硬膜外用药品
Intracerebral, intrathecal, and epidural drug products may directly interact with vital central nervous system (CNS) tissues that have a limited capacity for repair following insult, yet there is a paucity of data to characterize the potential toxicity of compounds directly administered into or in close proximity to neuronal tissue. In vitro data suggest chemically induced biological effects can occur in the very low parts per billion (ppb) range for some compounds with known neurotoxicity. Therefore, a compound-specific risk assessment should consider local concentration of observed leachables and the potential local toxicity concerns on neuronal tissue (e.g., neurons, astrocytes, glia, myelin) including an assessment of the potential for a local inflammatory response.
脑内、鞘内和硬膜外用药品可能直接与重要的中枢神经系统(CNS)组织相互作用,这些组织在受损后的修复能力有限,但关于直接给药至神经组织或其附近的化合物的潜在毒性,相关数据较为匮乏。体外数据表明,对于某些已知具有神经毒性的化合物,在极低的十亿分率(ppb)范围内即可产生化学诱导的生物学效应。因此,化合物特异性风险评估应考虑观察到的浸出物的局部浓度,以及对神经组织(如神经元、星形胶质细胞、神经胶质细胞、髓鞘)的潜在局部毒性担忧,包括对局部炎症反应潜力的评估。
6.4.3 Dermal Drug Products
6.4.3 皮肤用药品
With regard to any local toxicity effects, sensitization potential (see Section 6.4.4) is likely the most sensitive non-genotoxic endpoint when the leachable concerns a strong or extreme potency skin sensitizer. For High Potency Chemicals (HPC), a Dermal Sensitization Threshold (DST) of 1 µg/cm²/day has been derived. This threshold corresponds to 500 ppm in a dermal drug product, using the Cutaneous and Transcutaneous Concentration Limit (CTCL) calculation for conversion as described in ICH Q3D. Consequently, a local toxicity threshold corresponding to 500 ppm concentration in the product can be used for dermal products below which there is no need for local non-mutagenic toxicity evaluation including sensitization potential (See Table 1.).
关于局部毒性效应,当浸出物为强效或极强效皮肤致敏剂时,致敏潜力(见第 6.4.4 节)可能是最敏感的非遗传毒性终点。对于高 potency 化学品(HPC),皮肤致敏阈值(DST)为 1 µg/cm²/ 天。使用 ICH Q3D 中描述的皮肤和经皮浓度限值(CTCL)计算进行转换后,该阈值相当于皮肤用药品中的 500 ppm。因此,皮肤用药品可采用相当于产品中 500 ppm 浓度的局部毒性阈值,低于该阈值时,无需进行包括致敏潜力在内的局部非致突变性毒性评估(见表 1)。
6.4.4 Sensitization Potential
6.4.4 致敏潜力
Sensitizers are compounds that may trigger hypersensitivity reactions after repeated exposure. The concern for these compounds is dependent on the sensitization potential of the compound, the route of exposure and the susceptibility of the individual exposed. Different types of hypersensitivity with multiple modes of action have been described for various routes of exposure; however, validated prediction models exist for the dermal route only. This guidance addresses the risk for induction of sensitization potential and provides local toxicity thresholds for this risk where appropriate. If patients are sensitized to a compound, elicitation of sensitization reactions may occur at lower thresholds.
致敏剂是反复暴露后可能引发超敏反应的化合物。对这些化合物的担忧取决于化合物的致敏潜力、暴露途径以及暴露个体的易感性。针对不同暴露途径,已描述了多种作用模式的超敏反应类型;但目前仅针对皮肤途径存在经过验证的预测模型。本指南阐述了致敏潜力诱导的风险,并在适当情况下为此类风险提供局部毒性阈值。若患者对某一化合物致敏,在更低阈值下即可引发致敏反应。
Dermal exposure
皮肤暴露
Most data on sensitization potential have been obtained using the dermal route. Besides human data, in silico, in chemico, in vitro, and in vivo models have been developed and used to characterize the dermal sensitization potential of compounds. DSTs have been derived based on sensitization potency.1,2
大多数关于致敏潜力的数据来自皮肤途径。除人体数据外,已开发并使用计算机模拟、化学体外、体外和体内模型来表征化合物的皮肤致敏潜力。皮肤致敏阈值(DST)是基于致敏 potency 推导得出的 1,2。
Where an identified leachable is administered dermally below the DST for the relevant potency category, it can be concluded that no concern for dermal sensitization is expected, and no further action is required. If the DST is exceeded, available compound-specific data on sensitization potential should be evaluated. If no such data are available, or when these data raise concerns, risk mitigation measures need to be considered. These may include replacement of the component leaching the compound or reduction of the level of the leachable.
若已识别的浸出物通过皮肤给药时的剂量低于相应 potency 类别的 DST,则可得出结论预计不存在皮肤致敏担忧,无需采取进一步措施。若超过 DST,应评估现有的化合物特异性致敏潜力数据。若缺乏此类数据,或数据引发担忧,则需考虑风险缓解措施,可能包括更换浸出该化合物的组件或降低浸出物水平。
As transdermal drugs are applied to the skin as well, the same approach can be used to evaluate the risk for sensitization potential. For multi-day patches it is assumed that all leachables migrate within a day. A slower migration rate should be justified with data.
由于经皮给药药品也施用于皮肤,因此可采用相同方法评估其致敏潜力风险。对于多日贴剂,假设所有浸出物在一天内迁移完毕。迁移速率较慢的情况需有数据支持。
Inhalation exposure
吸入暴露
Knowledge of the respiratory sensitization potential of a compound is primarily from human data. Currently, suitable non-clinical models for respiratory sensitization are not established for safety risk assessment. The modes of action for dermal and respiratory sensitizers show commonalities, but also deviate, especially after T-cell activation. Consequently, dermal sensitization data should not be used to estimate the risk for respiratory sensitization and no threshold for respiratory sensitization can be provided.
关于化合物的呼吸道致敏潜力,相关知识主要来自人体数据。目前,尚未建立适用于安全风险评估的呼吸道致敏非临床模型。皮肤致敏剂和呼吸道致敏剂的作用模式虽有共性,但也存在差异,尤其是在 T 细胞激活后。因此,皮肤致敏数据不应用于评估呼吸道致敏风险,且无法提供呼吸道致敏的阈值。
The respiratory tract is very sensitive to compounds with sensitizing (and irritating) properties3. Therefore, any compound with structural elements that may suggest sensitizing potential or irritation should be evaluated (e.g. isocyanates, nitriles, styrenes, short-chain aldehydes). If a compound is considered to be an irritant or have sensitizing potential, patient risk should be assessed on a case-by-case basis after evaluating the available information for the specific compound. Additionally, available clinical data should be evaluated for evidence of any adverse effects. If no concern is identified for irritancy or sensitization, a systemic toxicity QT aligned with parenteral, as presented in Table 1, is considered appropriate.
呼吸道对具有致敏(和刺激)特性的化合物非常敏感 3。因此,任何具有可能提示致敏潜力或刺激性的结构元素的化合物都应进行评估(例如,异氰酸酯、腈类、苯乙烯、短链醛类)。若某一化合物被认为具有刺激性或致敏潜力,在评估该特定化合物的现有信息后,应逐案评估患者风险。此外,还应评估现有临床数据,以寻找任何不良效应的证据。若未发现刺激性或致敏性担忧,则采用与表 1 中胃肠外给药一致的全身毒性 QT 是适当的。
Parenteral Exposure
胃肠外暴露
Regarding potential risk for sensitization, a distinction should be made between the subcutaneous/intradermal route and the intravenous/intramuscular/intraperitoneal routes of exposure. For the subcutaneous route, the drug is administered in the vicinity of the same tissues and cells (i.e., Langerhans cells) that are pivotal in triggering dermal sensitization. Especially, when the leachable is not readily distributed and remains for more extended periods in the subcutis, the same modes of action may be activated. Consequently, available data on dermal sensitization potential can be informative when evaluating the sensitization potential for leachables that are administered subcutaneously. Likewise for products administered intradermally, dermal sensitization data may be of relevance. In contrast, dermally applied compounds need to penetrate the skin barrier first. To account for this difference a ten-fold lower threshold for subcutaneous and intradermal products as compared to dermal products is considered justified, i.e., 50 ppm instead of 500 ppm.
关于致敏的潜在风险,应区分皮下 / 皮内给药途径与静脉 / 肌内 / 腹腔内给药途径。对于皮下途径,药物施用于与触发皮肤致敏至关重要的相同组织和细胞(即朗格汉斯细胞)附近。特别是当浸出物不易分布且在皮下组织中长时间停留时,可能激活相同的作用模式。因此,在评估皮下给药浸出物的致敏潜力时,现有的皮肤致敏潜力数据可能具有参考价值。对于皮内给药产品,皮肤致敏数据也可能具有相关性。相比之下,经皮应用的化合物首先需要穿透皮肤屏障。为考虑这一差异,皮下和皮内产品的阈值被认为应比皮肤用产品低 10 倍是合理的,即 50 ppm 而非 500 ppm。
Several types of systemic hypersensitivity (Type I-IV) are known, each having different modes of action. Type IV is dependent on hapten formation and thus shares some mechanistic aspects with dermal sensitization. However, contrary to dermal application, intramuscular and intravenous administered substances are rapidly distributed systemically, and large amounts are required to activate the immune system and induce sensitization. Since leachables are present at low concentrations in drug products, it is considered unlikely that sensitization potential will be of concern for drugs administered via intravenous or intramuscular injection.
已知存在多种类型的全身性超敏反应(I-IV 型),每种类型都有不同的作用模式。IV 型依赖于半抗原的形成,因此与皮肤致敏存在一些机制上的共性。然而,与经皮给药不同,肌内和静脉给药的物质会迅速全身分布,且需要大量物质才能激活免疫系统并诱导致敏。由于浸出物在药品中的浓度较低,因此静脉或肌内注射给药的药品其浸出物的致敏潜力被认为不太可能引起担忧。
6.5 Considerations for ICH S9 Products
6.5 ICH S9 药品的考虑因素
For drug products within the scope of ICH S9, leachables should generally be identified according to the scientific principles outlined in Section 3 above. The safety risk assessment may be conducted according to the ‘Evaluation of Impurities’ Section in ICH S9. In this case, the TTC would not be applicable and the SCT would be defined by the QT. Risk assessment may be conducted with a focus on general safety for the intended patient population and is relevant for genotoxic APIs covered by ICH S9 Q&A, 2018.
对于 ICH S9 范围内的药品,浸出物的识别通常应遵循上文第 3 节所述的科学原则。安全风险评估可按照 ICH S9 中的 “杂质评估” 部分进行。在这种情况下,毒理学关注阈值(TTC)不适用,安全关注阈值(SCT)由确认阈值(QT)定义。风险评估可侧重于目标患者群体的总体安全性,且适用于 ICH S9 问答(2018 年)所涵盖的具有遗传毒性的原料药。
6.6 Content of Safety Assessment
6.6 安全评估的内容
A safety assessment should be conducted for observed Class 1 leachables, Class 2 leachables detected at levels above the relevant SCT, and Class 3 leachables when present at levels above 1.0 mg/day. The safety assessment should provide sufficient information to conclude on the acceptability of the anticipated patient exposure levels. Further details on the information to be considered and the methodology for deriving an acceptable exposure level is provided in Appendix 5.
应对观察到的 1 类浸出物、检测水平高于相关安全关注阈值(SCT)的 2 类浸出物,以及水平超过 1.0 mg / 天的 3 类浸出物进行安全评估。安全评估应提供充分信息,以得出关于预期患者暴露水平可接受性的结论。附录 5 提供了关于需考虑的信息以及推导可接受暴露水平的方法的更多细节。
7.GLOSSARY
术语表
Analytical Evaluation Threshold (AET):
The threshold above which an extractable or leachable should be identified, quantified, and reported for safety assessment.
分析评估阈值(AET)
高于该阈值的可提取物或浸出物应被识别、定量并报告,以进行安全评估。
Chemical characterization:
The process of obtaining chemical information about the composition of an item such as pharmaceutical packaging and a pharmaceutical manufacturing component.
化学表征
获取某一物品(如药品包装和药品生产组件)成分的化学信息的过程。
Component:
A single item, composed of one or more materials of construction, that serves a single purpose or performs a single and specific task.
组件
由一种或多种构造材料组成的单个物品,用于实现单一目的或执行单一特定任务。
Extraction:
The chemical or physical process of transferring constituents of a test article into an extraction medium.
提取
将测试样品中的成分转移到提取介质中的化学或物理过程。
Critical quality attribute:
A physical, chemical, biological or microbiological property or characteristic that should be within an appropriate limit, range, or distribution to ensure the desired product quality.
关键质量属性
为确保达到预期的产品质量,应处于适当限度、范围或分布内的物理、化学、生物学或微生物学性质或特征。
Drug product:
The dosage form in the final immediate packaging intended for marketing.
药品
处于最终直接包装中、拟用于销售的剂型。
Drug substance:
The unformulated active pharmaceutical ingredient that may subsequently be formulated with excipients to produce the dosage form (or drug product).
原料药
未配制的活性药物成分,后续可与辅料一起配制成剂型(或药品)。
Extractables Profile:
Qualitative or semi-quantitative/quantitative accounting of the extractables present in an extract.
可提取物特征
对可提取物中存在的可提取物的定性或半定量 / 定量描述。
Leachables Profile:
Qualitative and/or quantitative accounting of the leachables present in a drug product.
浸出物特征
对药品中存在的浸出物的定性和 / 或定量描述。
Lifecycle:
All phases in the life of a product from the initial development through marketing until the product’s discontinuation
生命周期
产品从初始开发到上市直至停产的所有阶段。
Lowest-Observed (Adverse) Effect Level (LO(A)EL):
The lowest dose of substance in a study or group of studies that produces biologically significant increases in frequency or severity of any (adverse) effects in the exposed humans or animals.
最低观察到(不良)效应水平(LO (A) EL)
在一项或一组研究中,使暴露的人或动物出现任何(不良)效应的频率或严重程度产生生物学意义上显著增加的物质最低剂量。
Read-across:
A technique for predicting endpoint information for one substance by using data from the same endpoint from (an)other structurally-related substance(s).
交叉参照
通过使用来自一种或多种结构相关物质的相同终点数据,来预测某一物质的终点信息的技术。
Margin of Safety:
A correlation between the PDE of the specific leachable and actual patient intake based on the daily dose.
安全边际
特定浸出物的允许日暴露量(PDE)与基于日剂量的患者实际摄入量之间的关联。
Materials of construction:
Individual materials used to construct a packaging or manufacturing component or system.
构造材料
用于制造包装或生产组件或系统的各种单独材料。
New drug product:
A pharmaceutical product type, for example, tablet, capsule, solution, cream, which has not previously been registered in a region or Member State, and which contains a drug ingredient generally, but not necessarily, in association with excipients.
新药品
一种药品类型(例如片剂、胶囊剂、溶液剂、乳膏剂),此前未在某一地区或成员国注册,通常含有药物成分(但不一定与辅料结合)。
No Observed (Adverse) Effect Level (NO(A)EL):
The highest concentration or amount of a leachable or extractable that does not cause any statistically or biologically significant (adverse) effects in the exposed population compared to a control group.
未观察到(不良)效应水平(NO (A) EL)
与对照组相比,在暴露群体中不会引起任何统计学或生物学意义上显著(不良)效应的浸出物或可提取物的最高浓度或量。
Permitted Daily Exposure (PDE):
The maximum acceptable intake per day of a leachable in pharmaceutical products per day (for a lifetime).
允许日暴露量(PDE)
药品中某一浸出物的每日最大可接受摄入量(终生)。
Point of Departure (PoD):
Starting point in the calculation of PDE of leachables; it can be derived from the human dose or appropriate animal study.
起始点(PoD)
计算浸出物允许日暴露量(PDE)的起始点;可从人体剂量或适当的动物研究中得出。
Qualification Threshold (QT):
Threshold above which a leachable should be qualified for potential non-mutagenic toxicity unless the leachable is identified as being Class 1.
确认阈值(QT)
高于该阈值的浸出物应进行潜在非致突变性毒性确认,除非该浸出物被确定为 1 类浸出物。
Safety Concern Threshold (SCT):
Threshold at or below which a leachable would have a dose so low as to present negligible safety concerns from mutagenic and non-mutagenic toxic effects unless the leachable is identified as being a leachable of high concern.
安全关注阈值(SCT)
在该阈值或以下时,浸出物的剂量极低,以至于其致突变性和非致突变性毒性效应带来的安全担忧可忽略不计,除非该浸出物被确定为高关注浸出物。
Simulated Drug Product:
Matrix or solvent that mimics closely the leaching characteristics of the drug product formulation with respect to leaching propensity and solubility of leachables.
模拟药品
在浸出倾向和浸出物溶解度方面,密切模拟药品配方浸出特性的基质或溶剂。
Substance (Compound, Chemical, Chemical Entity):
An association of different elements or chemical entities which have a definite chemical composition and distinct chemical properties.
物质(化合物、化学品、化学实体)
由不同元素或化学实体结合而成,具有确定的化学组成和独特的化学性质。
System:
The sum of individual components (or assemblies) which together perform a specific function, such as manufacturing, delivery or storage/packaging.
系统
共同执行特定功能(如生产、给药或储存 / 包装)的各个组件(或组件集合)的总和。
Threshold of Toxicological Concern (TTC):
Threshold at or below which a leachable is not considered for safety assessment for mutagenic effects as described in ICH M7.
毒理学关注阈值(TTC)
如 ICH M7 所述,在该阈值或以下时,浸出物无需进行致突变性效应的安全评估。
8.REFERENCES
参考文献
International Conference on Harmonisation (2006). Q3A(R2): Impurities in New Drug Substances.
International Conference on Harmonisation (2006). Q3B(R2): Impurities in New Drug Products.
International Council for Harmonisation (2024). Q3C(R9): Guideline for Residual Solvents.
International Council for Harmonisation (2022). Q3D(R2): Guideline for Elemental Impurities.
International Council for Harmonisation (2023). M7(R2): Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk.
International Council for Harmonisation (2023). Q9(R1): Quality Risk Management.
International Conference on Harmonisation (2009). S9: Nonclinical Evaluation for Anticancer Pharmaceuticals.
International Council for Harmonisation (2018). S9: Nonclinical Evaluation for Anticancer Pharmaceuticals Questions and Answers.
Chilton ML, Api AM, Foster RS, Gerberick GF, Lavelle M, Macmillan DS, et al. Updating the Dermal Sensitisation Thresholds using an expanded dataset and an in silico expert system. Regul Toxicol Pharmacol. 2022; Aug;133:105200,
Parris P, Whelan G, Burild A, Whritenour J, Bruen U, Bercu J, et al. Sensitization Assessment of Extractables and Leachables in Pharmaceuticals: ELSIE Database Analysis. PDA J Pharm Sci Technol. 2024 Aug 23;78(4):399-444.
Ball D, Blanchard J, Jacobson-Kram D, McClellan RO, McGovern T, Norwood DL, et al. Development of Safety Qualification Thresholds and Their Use in Orally Inhaled and Nasal Drug Product Evaluation. Toxicol Sci. 2007 Jun;97(2):226-36.
Appendix 1: Typical workflows for E&L risk assessment and risk control
附录 1: 可提取物和浸出物(E&L)风险评估及风险控制的典型工作流程
The following diagrams illustrate typical workflows for E&L overall risk assessment and risk control, for component qualifications for manufacturing components/systems packaging (Figure 4) and packaging and delivery device components/systems (Figure 5). Typically for manufacturing components/systems and under most circumstances for packing systems, a safety assessment of leachable studies considering worst case conditions is expected. However, under certain low risk circumstances, alternative approaches can be proposed. In all instances, similar to the examples given in Table A.1.1 and Table A.1.2 and where other low-risk scenarios could occur, the approach taken should be justified (see Table A.1.1 and Table A.1.2). Overall, it is expected that the extent of data requirements and subsequent quality and safety assessment is commensurate with the overall level of risk.
以下图表展示了 E&L 整体风险评估及风险控制的典型工作流程,涉及生产组件 / 系统包装的组件确认(图 4)以及包装和给药装置组件 / 系统的组件确认(图 5)。通常对于生产组件 / 系统,以及在大多数情况下对于包装系统,需要结合最坏情况对浸出物研究进行安全性评估。然而,在某些低风险情况下,可以提出替代方法。在所有情况下,与表 A.1.1 和表 A.1.2 中给出的示例类似,以及在可能出现其他低风险场景时,所采用的方法都应提供正当理由(参见表 A.1.1 和表 A.1.2)。总体而言,数据要求的范围以及后续的质量和安全性评估应与整体风险水平相匹配。
Figure 4: Typical workflow for E&L assessment related risk identification and mitigation for manufacturing components/systems
图 4:与生产组件 / 系统相关的可提取物与可浸出物(E&L)评估的典型工作流程 — 风险识别与缓解
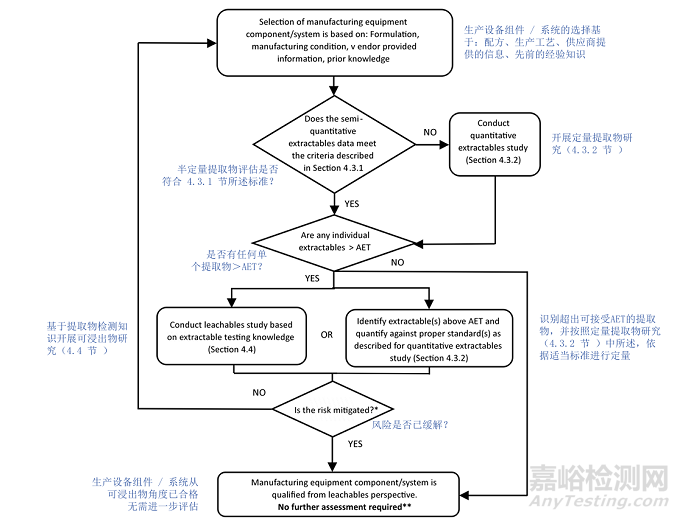
Refer to Section 4.3 for method qualification and chemical identification expectations as well as scenarios where a leachable study is recommended.
有关方法确认、化学物质识别的要求以及建议进行浸出物研究的场景,请参见第 4.3 节。
Amount of extractable(s) or leachable(s) are below the applicable safety threshold for each compound.
每种化合物的可提取物或浸出物量低于适用的安全阈值。
** For manufacturing process employing multiple components constructed with the same or similar material, cumulative leachables risk should be assessed for the final drug product (see Section 3.4.1).
** 对于使用多个由相同或相似材料制成的组件的生产工艺,应评估最终药品中浸出物的累积风险(参见第 3.4.1 节)。
Figure 5: Typical workflow for E&L assessment related risk identification and mitigation for packaging and delivery device components
图 5:与包装及给药装置组件相关的可提取物与可浸出物(E&L)评估的典型工作流程 — 风险识别与缓解
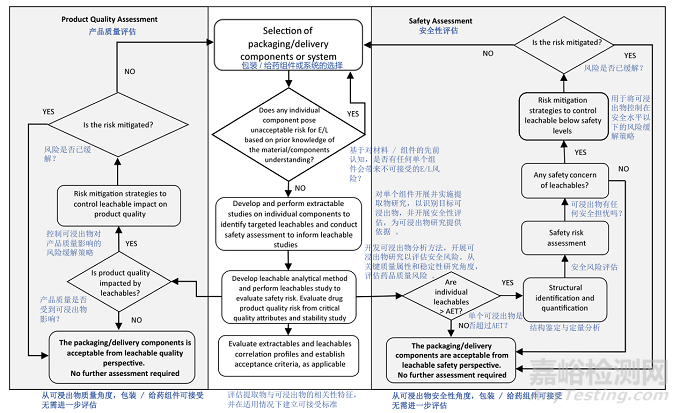
Table A.1.1: Manufacturing Equipment Components/Systems Scenarios
表 A.1.1:生产设备组件 / 系统场景
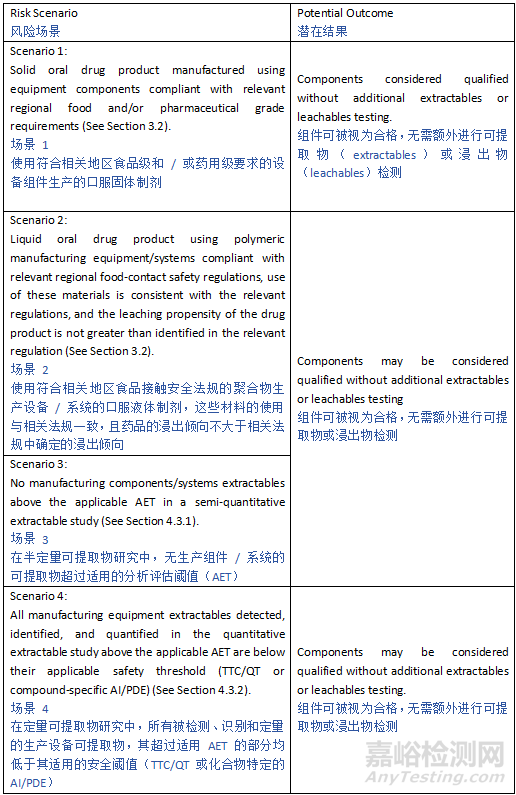
In general, comprehensive extractable and leachable data should be provided for all primary packaging components/systems and delivery device components.
一般而言,应为所有初级包装组件 / 系统和给药装置组件提供全面的可提取物和浸出物数据。
However, for overall low - risk scenarios (see Figure 2, Section 3.2) an abbreviated data package that includes a quantitative extractables study may be adequate with justification.
然而,对于整体低风险场景(见第 3.2 节图 2),经合理说明后,包含定量可提取物研究的简化数据包可能就足够。
See Section 3.4 for situations where a leachable study should be conducted to address the specific concerns and demonstrate acceptability of the components.
若需开展浸出物研究以解决特定问题并证明组件的可接受性,相关情形请见第 3.4 节 。
Table A.1.2: Examples For Abbreviated Data Package for Packaging and Delivery Device Components
表 A.1.2:包装及给药装置组件简化数据包示例
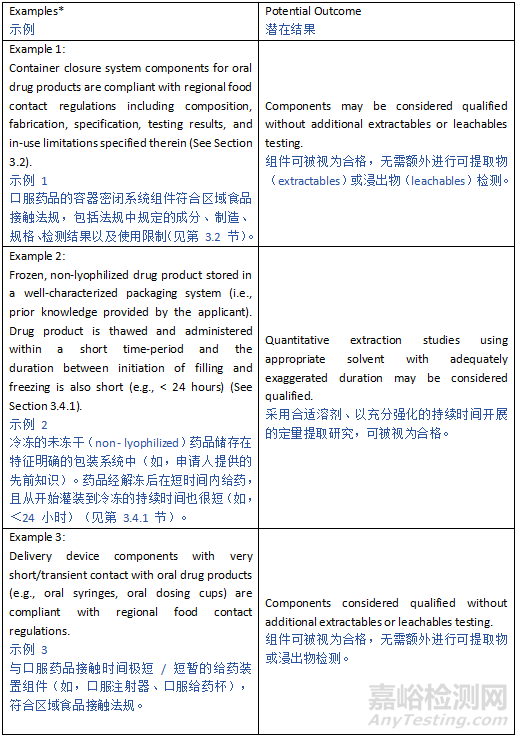
Note 1 for Table A.1.1 and Table A.1.2:
表 A.1.1 和表 A.1.2 的注释 1
Refer to section 4.3 for recommendations for extractable and leachable study, as appropriate.
如需可提取物和浸出物研究相关建议,酌情参考第 4.3 节 。
Refer to section 3.5 for recommendation for appropriate documentation and compliance, as appropriate.
如需恰当文件记录和合规性相关建议,酌情参考第 3.5 节 。
*If no or few extractables are detected above the AET, and below their applicable safety threshold (such as Class 3 leachables; See Section 6), in conjunction with prior knowledge an abbreviated data package may be warranted with adequate justification. When an abbreviated data package is proposed, communications with relevant regional Regulatory Agency/Health Authority is recommended to align on approach.
* 若检测到高于分析评估阈值(AET)但低于其适用安全阈值(如 3 类浸出物;见第 6 节 )的可提取物数量为零或很少,结合先前知识,经充分论证后,采用简化数据包可能是合理的。若提议采用简化数据包,建议与相关地区的监管机构 / 卫生当局沟通,以在方法上达成一致 。
Appendix 2: Types of Studies
附录 2:研究类型
Table A.2.1: Summary of Extractable, Leachable and Simulated Leachable Studies
表 A.2.1:可提取物、可浸出物及模拟可浸出物研究概述

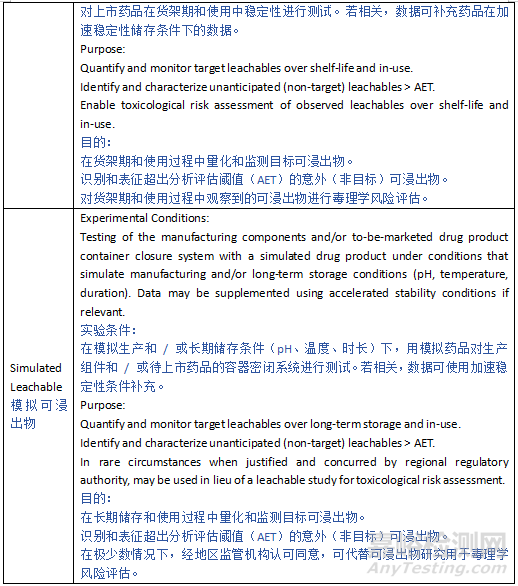
Refer to Section 4.3 for detailed recommendations for extractable and leachable study, as appropriate.
酌情参考第 4.3 节,获取有关可提取物和可浸出物研究的详细建议。
Appendix 3: AET Calculations
附录 3: 分析评估阈值(AET)计算
Each of the examples provided are based upon using the applicable SCT (µg/day) for the drug product. In some instances, an alternative starting point may be pertinent (such as for a potential Class 1 leachable). In all calculations, worst-case assumptions such as maximum approved dosing of the drug product should be assumed. Common examples for both extractables and leachables studies are provided. Calculation of the AET should clearly indicate what the units are and how the calculation was performed. Regardless of the units used to express the AET, the final value for a given study should always equate to the same patient exposure level (i.e., the SCT multiplied by the analytical uncertainty factor [UF]).
所提供的每个示例均基于药品适用的安全关注阈值(SCT,单位:微克/天 )。在某些情况下,替代起始点可能适用(比如针对潜在的1类可浸出物 )。在所有计算中,均应采用最坏情况假设,如假设药品的最大获批给药剂量 。提供了可提取物和可浸出物研究的常见示例。分析评估阈值(AET)的计算应明确说明单位是什么以及计算是如何进行的 。无论用于表示AET的单位如何,特定研究的最终值应始终等同于相同的患者暴露水平(即SCT乘以分析不确定因子(UF) )。
Maximum Daily Dose (MDD) and Safety Concern Threshold (SCT)
最大日剂量(MDD)与安全关注阈值(SCT)
For each product the calculation of the AET should be based on the MDD.The MDD is the maximum approved dose of a drug administered in a single day.
对于每种产品,AET 的计算均应基于 MDD 。MDD 是指药品在一日内给药的最大获批剂量 。
To determine the SCT, both the TTC and QT should be considered, as indicated in Table 1.The lowest of these values determines the SCT.
如表格 1 所示,确定 SCT 时,应同时考量毒理学关注阈值(TTC)和定量阈值(QT) 。这些数值中的最小值决定 SCT 。
Intermittent Dosing
间歇性给药
If a drug is not administered every day, for derivation of the applicable TTC ICH M7 is followed (e.g., when total number of dosing days is ≤30, the TTC = 120 µg).
若药品并非每日给药,推导适用的 TTC 时应遵循 ICH M7(例如,当给药总天数≤30 天,TTC = 120 微克 )。
For derivation of the QT, when total number of dosing days is ≤30 days or the dosing frequency is once per month or less, the ≤ 1 month QT can be used.
推导 QT 时,若给药总天数≤30 天,或给药频率为每月一次或更低,可采用≤1 个月对应的 QT 。
Multi - day Products
多日使用产品
For products that are applied and may remain in place for multiple days (e.g. multi - day patches, depot injections, implants), the applicable TTC is defined by the total duration of treatment.For mutagenic impurities, per ICH M7 an average daily exposure should be used.For non - mutagenic leachable, the default assumption is that all leachables migrate within a day.In this case, the applicable QT is defined by the total number of applications.A slower migration rate would decrease the daily dose to a non - mutagenic leachable but increase the number of dosing days.A slower migration rate should be justified with data.
对于施用后可能留存多日的产品(如多日贴剂、缓释注射剂、植入物),适用的 TTC 由治疗总时长界定 。对于致突变性杂质,依据 ICH M7,应采用平均每日暴露量 。对于非致突变性可浸出物,默认假设是所有可浸出物在一日内迁移 。在此情形下,适用的 QT 由施用总次数界定 。迁移速率较慢会降低非致突变性可浸出物的日剂量,但会增加给药天数 。迁移速率较慢的情况需以数据为依据进行说明 。
Example AET Calculations
示例分析评估阈值(AET)计算
Extractable Scenario 1: Filter used as part of a manufacturing process for a liquid drug product
可提取物场景 1:用于液体制药生产工艺的过滤器
(1)AET (µg/filter) = SCT (µg/day) × UF × Doses per drug product batch* ÷ Filters/batch
(1)AET(微克 / 过滤器)= 安全关注阈值(SCT,微克 / 天)× 不确定因子(UF)× 每批药品的剂量数 * ÷ 每批所用过滤器数量
(2)AET (µg/g filter) = AET (µg/filter) ÷ Weight (g)/filter
(2)AET(微克 / 克过滤器)= AET(微克 / 过滤器)÷ 过滤器重量(克 / 个)
(3)AET (µg/mL extraction solvent) = AET (µg/filter) ÷ Extraction solvent (mL)/filter
(3)AET(微克 / 毫升提取溶剂)= AET(微克 / 过滤器)÷ 每个过滤器所用提取溶剂体积(毫升)
(4)AET (µg/cm²) = AET (µg/filter) ÷ Contact surface area (cm²)/filter
(4)AET(微克 / 平方厘米)= AET(微克 / 过滤器)÷ 每个过滤器的接触表面积(平方厘米)
*The MDD administered in a single day and the minimum potential batch size should be used to determine the number of doses per drug product batch (i.e., the worst-case scenario). Thus, if the maximum approved dose given in a single day is 100 mg (= 0.1 g) and the minimum potential batch size in 1 kg (= 1000 g), the doses per drug product batch is 1000 g/batch ÷ 0.1 g/dose = 10,000 doses per drug product batch.
* 应使用单日最大日剂量(MDD)和最小潜在批量来确定每批药品的剂量数(即最坏情况)。因此,若单日最大获批剂量为 100 毫克(=0.1 克),最小潜在批量为 1 千克(=1000 克),则每批药品的剂量数为 1000 克 / 批 ÷ 0.1 克 / 剂量 = 10,000 剂量 / 批。
Extractable Scenario 2: Rubber vial stopper as part of CCS for a liquid drug product
可提取物场景 2:作为液体制品容器密闭系统(CCS)组成部分的橡胶瓶塞
(1)AET (µg/stopper) = SCT (µg/day) × UF × Volume/vial (mL/stopper) ÷ Maximum dose in a day (mL)*
(1)AET(微克 / 瓶塞)= 安全关注阈值(SCT,微克 / 天)× 不确定因子(UF)× 每瓶体积(毫升 / 瓶塞)÷ 单日最大剂量(毫升)*
(2)AET (µg/g stopper) = AET (µg/stopper) ÷ Stopper weight (g)
(2)AET(微克 / 克瓶塞)= AET(微克 / 瓶塞)÷ 瓶塞重量(克)
(3)AET (µg/mL extraction solvent) = AET (µg/stopper) ÷ Extraction solvent (mL)/Stopper
(3)AET(微克 / 毫升提取溶剂)= AET(微克 / 瓶塞)÷ 每个瓶塞所用提取溶剂体积(毫升)
(4)AET (µg/mL extraction solvent) = AET (µg/g stopper) ÷ Extraction solvent (mL)/gram of Stopper
(4)AET(微克 / 毫升提取溶剂)= AET(微克 / 克瓶塞)÷ 每克瓶塞所用提取溶剂体积(毫升)
*The maximum approved volumetric dose administered in a single day should be used (i.e., the worst-case scenario). If dosing is described on a mass basis (e.g., mg/day), it should be converted to a volume (mL) based upon the concentration of the active ingredient. Thus, if the maximum approved dose given in a single day is 100 mg (= 0.1 g) and the concentration of the drug product is 10 mg/mL, the maximum dose in a day for the calculation is 100 mg ÷ 10 mg/mL = 10 mL.
* 应使用单日最大获批体积剂量(即最坏情况)。若剂量以质量单位描述(如毫克 / 天),应根据活性成分浓度转换为体积(毫升)。因此,若单日最大获批剂量为 100 毫克(=0.1 克),药品浓度为 10 毫克 / 毫升,则计算所用的单日最大剂量为 100 毫克 ÷ 10 毫克 / 毫升 = 10 毫升。
Leachable Scenario 1: Leachables for manufacturing equipment for liquid drug product
可浸出物场景 1:液体制药生产设备的可浸出物
(1)AET (µg/batch) = SCT (µg/day) × UF × Doses per drug product batch*
(1)AET(微克 / 批)= 安全关注阈值(SCT,微克 / 天)× 不确定因子(UF)× 每批药品的剂量数 *
(2)AET (µg/mL drug product) = SCT (µg/day) × UF ÷ Maximum dose in a day (mL)
(2)AET(微克 / 毫升药品)= 安全关注阈值(SCT,微克 / 天)× 不确定因子(UF)÷ 单日最大剂量(毫升)
*The MDD administered in a single day and the minimum potential batch size should be used to determine the number of doses per drug product batch (i.e., the worst-case scenario). Thus, if the maximum approved dose given in a single day is 5 mL and the minimum potential batch size in 10 L (= 10,000 mL), the doses per drug product batch is 10,000 mL/batch ÷ 5 mL/dose = 2,000 doses per drug product batch.
* 应使用单日最大日剂量(MDD)和最小潜在批量来确定每批药品的剂量数(即最坏情况)。因此,若单日最大获批剂量为 5 毫升,最小潜在批量为 10 升(=10,000 毫升),则每批药品的剂量数为 10,000 毫升 / 批 ÷ 5 毫升 / 剂量 = 2,000 剂量 / 批。
Leachable Scenario 2: Leachables for a prefilled syringe (PFS)
可浸出物场景 2:预填充注射器(PFS)的可浸出物
(1)AET (µg/mL drug product) = SCT (µg/day) × UF ÷ Maximum dose in a day (mL)*
(1)AET(微克 / 毫升药品)= 安全关注阈值(SCT,微克 / 天)× 不确定因子(UF)÷ 单日最大剂量(毫升)*
(2)AET (µg/PFS) = AET (µg/mL drug product) × Volume per PFS (mL)
(2)AET(微克 / 预填充注射器)= AET(微克 / 毫升药品)× 每个预填充注射器的体积(毫升)
*The maximum approved volumetric dose administered in a single day should be used (i.e., the worst-case scenario). If dosing is described on a mass basis (e.g., mg/day), it should be converted to a volume (mL) based upon the concentration of the active ingredient. Thus, if the maximum approved dose given in a single day is 10 mg and the concentration of the drug product is 10 mg/mL, the maximum dose in a day for the calculation is 10 mg ÷ 10 mg/mL = 1 mL.
* 应使用单日最大获批体积剂量(即最坏情况)。若剂量以质量单位描述(如毫克 / 天),应根据活性成分浓度转换为体积(毫升)。因此,若单日最大获批剂量为 10 毫克,药品浓度为 10 毫克 / 毫升,则计算所用的单日最大剂量为 10 毫克 ÷ 10 毫克 / 毫升 = 1 毫升。
Appendix 4: Potency Classes for Leachables
附录 4:可浸出物的效力等级
The chemical nature of potential leachable compounds is varied as are their safety databases. In order to remain patient protective while maintaining a practical approach to setting safety thresholds, a leachables classification scheme has been developed, in addition to the thresholds applied in the guideline.
潜在可浸出物的化学性质各异,其安全性数据库也各不相同。为在保护患者的同时,以切实可行的方式设定安全阈值,除本指南中应用的阈值外,还制定了可浸出物分类体系。
The classification scheme is based on systemic effects and is broadly applicable to all routes of administration. However, the concentration thresholds applicable to drug products with specific routes of administration as indicated in Section 6.1 Table 1 are not impacted by this classification scheme. As such, the default concentration thresholds for potential local effects of a leachable are the same regardless of leachable class.
该分类体系基于全身效应,广泛适用于所有给药途径。但第 6.1 节表 1 中所列特定给药途径药品的浓度阈值不受此分类体系影响。因此,无论可浸出物属于哪一等级,其潜在局部效应的默认浓度阈值均相同。
Class 1 leachables are generally those compounds for which the thresholds for mutagenic and systemic effects as described in this guideline have not been demonstrated to be sufficiently patient protective. Thus, for Class 1 leachables an acceptable exposure level should be established on a compound-specific basis.
1 类可浸出物通常指本指南中所述的致突变效应和全身效应阈值未被证明能充分保护患者的化合物。因此,1 类可浸出物的可接受暴露水平应根据具体化合物确定。
Class 1 includes: ICH M7 cohort of concern compounds, ICH M7 Class 1 compounds with an AI < 1.5 µg/day, and non-mutagenic leachables with a derived Permitted Daily Exposure (PDE) following the methodology described in Appendix 5 for which the established QT values may not be protective of patient safety (see Appendix 6).
1 类可浸出物包括:ICH M7 关注组化合物、每日可接受摄入量(AI)<1.5 微克的 ICH M7 1 类化合物,以及采用附录 5 所述方法得出允许日暴露量(PDE)、且既定定量阈值(QT)可能无法保护患者安全的非致突变性可浸出物(见附录 6)。
Class 2 is the default leachable classification and includes compounds for which the chronic parenteral administration thresholds for mutagenicity (TTC) and systemic toxicity (QT), as described in this guideline, are considered to be sufficiently patient protective.
2 类是可浸出物的默认分类,包括本指南所述的慢性肠外给药致突变性阈值(TTC)和全身毒性阈值(QT)被认为能充分保护患者的化合物。
This includes all compounds for which a PDE was not specifically listed in this guideline.
这包括本指南中未具体列出允许日暴露量(PDE)的所有化合物。
Class 3 leachables are compounds established to have relatively low potency for systemic toxicity with derived chronic parenteral PDE in excess of the levels at which leachables are typically observed. Class 3 leachables would not require further safety qualification if observed at daily exposure levels < 1.0 mg/day.
3 类可浸出物是经确认具有相对较低全身毒性效力的化合物,其得出的慢性肠外给药允许日暴露量(PDE)超过可浸出物通常观察到的水平。若 3 类可浸出物的每日暴露水平 < 1.0 毫克 / 天,则无需进一步的安全性确认。
A summary of these leachables classes is provided in Table A.4.1, below.
这些可浸出物等级的概述详见下文表 A.4.1。
Leachable levels greater than identified in Table A.4.1 should be scientifically justified as described in Appendix 5.
超过表 A.4.1 中规定水平的可浸出物,应按照附录 5 所述进行科学论证。
Table A.4.1: Potency Classes for Leachables
表 A.4.1:可浸出物的效力等级
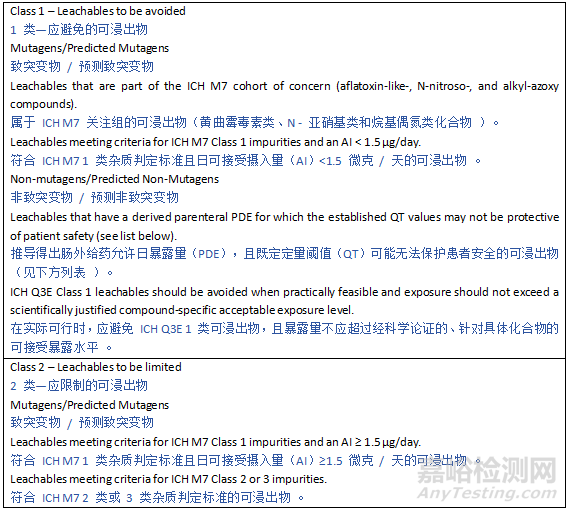

Class 1 Leachables to be avoided
1 类应避免的可浸出物
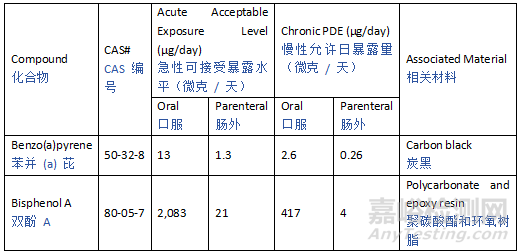
Class 3 Leachables With Relatively Low Toxic Potential (Chronic Parenteral PDE ≥ 1 mg/day). Monographs In Supporting Documents.
3 类毒性潜力相对较低的可浸出物(慢性肠外给药允许日暴露量≥1 毫克 / 天 )。支持性文件中的各论 。

Appendix 5: Methods for Establishing Exposure Limits
附录 5:暴露限值的确定方法
Background
背景
For Class 1 leachables and Class 2/3 leachables exceeding their applicable safety threshold as defined in this guideline, further safety assessment is performed to establish the potential risk associated with exposure to these leachables when a patient is administered a specific drug product.
对于 1 类可浸出物以及超出本指南所定义适用安全阈值的 2 类 / 3 类可浸出物,当患者使用特定药品时,需开展进一步安全评估,以确定接触这些可浸出物带来的潜在风险。
Permitted Daily Exposure (PDE) values intended to support safe exposure to a compound in any drug product are not currently established for the vast majority of potential leachables.
目前,对于绝大多数潜在可浸出物,尚未确立能支持其在任意药品中安全暴露的允许日暴露量(PDE)数值。
Furthermore, due to the varied nature of currently available drug products and the complexity of extractables and leachables safety risk assessment, a one size fits all approach, such as an established PDE, is not always most pertinent.
此外,鉴于现有药品的多样性以及可提取物和可浸出物安全风险评估的复杂性,“一刀切” 的方法(如既定的 PDE )并非总是最恰当的。
Although the focus of this guideline is not on the generation of acceptable exposure levels for individual compounds, the need for compound-specific limits on a product-by-product basis may commonly arise.
尽管本指南重点并非针对单个化合物生成可接受暴露水平,但按产品逐个确定化合物特定限值的需求经常出现。
Therefore, this appendix provides guidance to appropriately establishing the safety of leachables for a variety of drug product types and administration scenarios using a risk-based approach.
因此,本附录提供指导,以基于风险的方法,针对各类药品类型和给药场景,合理确立可浸出物的安全性。
The extent of the information considered sufficient to conclude on the acceptability of potential patient exposure levels for a leachable may vary extensively and there are multiple methodologies which may be employed to establish this acceptability.
判定可浸出物潜在患者暴露水平是否可接受所需的信息详尽程度差异极大,且有多种方法可用于确立这种可接受性。
The most straight-forward methodology is to employ already established safe exposure levels which have conservatively assumed worst scenarios.
最直接的方法是采用已确立的、保守假设最坏情形的安全暴露水平。
Thus, when there is an established PDE in an available ICH guidance (e.g., Q3C or M7) it is sufficient to refer to this value assuming all requisite considerations are met.
因此,若现有 ICH 指导原则(如 Q3C 或 M7 )中已确立 PDE,且满足所有必要考量,直接引用该数值即可。
Alternatively, an acceptable exposure derived using similar methodologies and scientific principles as previously established in such guidelines may be deemed more applicable or necessary.
或者,采用与这些指导原则中先前确立的类似方法和科学原理推导得出的可接受暴露水平,可能被认为更适用或更有必要。
In still other scenarios, the dose ratio between a well-defined, supported and justified NOAEL and the anticipated patient exposure may be so large (e.g., >10,000) that a detailed derivation may not be necessary.
在其他情形下,清晰界定、有依据且合理的未观察到不良反应水平(NOAEL )与预期患者暴露量的剂量比可能极大(如 > 10,000 ),此时可能无需详细推导。
Though in certain circumstances, in vitro and/or in vivo studies (as a last resort) may be deemed necessary to establish an acceptable exposure level, scientific justification (if applicable) via available in silico analyses and through read across to similar compounds (i.e., surrogate compound[s]) is encouraged to establish acceptable exposure levels.
尽管在某些情况下,体外和 / 或体内研究(作为最后手段 )可能被认为是确立可接受暴露水平所必需的,但仍鼓励通过现有计算机模拟分析以及关联到类似化合物(即替代化合物 )的科学论证(如适用 )来确立可接受暴露水平。
Although a variety of in silico toxicological tools are available, mutagenicity is the only toxicological endpoint for which such an appropriately conducted evaluation is currently well-established for stand-alone use in lieu of biological data within the context of this guideline (see ICH M7).
尽管有多种计算机毒理学工具可用,但在本指南范围内,致突变性是目前唯一一种经恰当开展的评估、已确立可单独用于替代生物学数据的毒理学终点(见 ICH M7 )。
However, with appropriate scientific justification, predictions of other toxicological endpoints using in silico, in vitro, or in vivo models should be incorporated into the safety risk assessment to supplement any existing data in a weight-of-evidence risk-based approach.
不过,在有合理科学论证的情况下,使用计算机模拟、体外或体内模型对其他毒理学终点的预测,应纳入安全风险评估,以基于证据权重的风险评估方法补充现有数据。
Within each of these categories, greater priority should be given to data from validated models that account for the relevant exposure route(s).
在上述每类方法中,应优先考虑经验证模型得出的数据,且这些模型需涵盖相关暴露途径。
Due to the limited nature or even lack of toxicological datasets for a large number of potential leachables, a read-across approach may also be incorporated.
由于大量潜在可浸出物的毒理学数据集有限甚至缺失,也可采用关联(read-across)方法。
In a read-across approach, toxicological data for a surrogate compound (or multiple surrogates) with pertinent toxicological data are used to support the safety assessment of a leachable of interest either as part of a weight-of-evidence approach or in lieu of data for the leachable of interest when none is available.
在关联方法中,具有相关毒理学数据的替代化合物(或多个替代物)的毒理学数据,可用于支持目标可浸出物的安全评估—— 作为证据权重方法的一部分,或在目标可浸出物无数据时替代其数据。
Safety assessments incorporating a surrogate compound should provide clear justification for the selection of the surrogate(s).
纳入替代化合物的安全评估,应对替代物的选择提供清晰论证。
There are various attributes that should be considered (if known) during the selection of a suitable surrogate, including mode of action, the principal toxicophore and surrounding chemical environment (e.g., presence of functional groups that may impact biological activity), overall structural similarity, toxicokinetic properties, physicochemical properties (e.g., polarity, solubility, ionizability, and molecular weight).
选择合适替代物时(如有已知信息),应考虑多种属性,包括作用模式、主要毒效基团及周边化学环境(如可能影响生物活性的官能团存在情况)、整体结构相似性、毒代动力学性质、理化性质(如极性、溶解度、离子化程度和分子量)。
When properly justified, in silico tools and data from NAMs may be used to support the selection of surrogates and inform the read-across approach, but the above-mentioned criteria need to be considered.
在恰当论证后,计算机模拟工具和非动物实验方法(NAMs )的数据可用于支持替代物选择并为关联方法提供信息,但仍需考虑上述标准。
How a surrogate is incorporated into the safety assessment for the leachable of interest should be scientifically justified.
替代物如何纳入目标可浸出物的安全评估,应进行科学论证。
Potential uncertainties related to the read-across approach should also be indicated and appropriately accounted for, such as when using for an acceptable exposure level determination (see F7 discussion below).
与关联方法相关的潜在不确定性也应指明并合理考量,比如用于确定可接受暴露水平时(见下文 F7 讨论 )。
Data to be Evaluated and Incorporated into the Safety Assessment
需评估并纳入安全评估的数据
In order to establish the safety of a leachable in a specific drug product, a thorough safety assessment of the compound should be provided.
为确定特定药品中某可浸出物的安全性,应对该化合物开展全面的安全评估。
Data elements to be included (where data are available) are listed below. The relevance and quality of these datasets should also be assessed.
以下列出需纳入的各类数据(如有相关数据)。还应评估这些数据集的相关性与质量。
As noted above, any use of surrogate compound data with in silico analyses should also be incorporated into the safety assessment and justified.
如前文所述,使用替代化合物数据结合计算机模拟分析的情况,也应纳入安全评估并进行论证。
Additionally, if several observed leachables are grouped together for evaluation, the details and justification of this grouping should be included.
此外,若将多种已观测到的可浸出物分组进行评估,需说明分组细节及理由。
Pharmacological/Biological Data
药理 / 生物学数据
Consider available in vivo or in vitro data that suggest the potential for biological effects that could impact the overall safety assessment (e.g., endocrine disruption, anticholinergic activity).
考量现有体内或体外数据,这些数据若提示可能存在影响整体安全评估的生物学效应(如内分泌干扰、抗胆碱能活性),需加以关注。
Toxicokinetics (TK)
毒代动力学(TK)
Assess and summarize data relevant to the drug product’s route of administration
评估并总结与药品给药途径相关的数据
Consider potential differences between absorption and bioavailability, especially when route-to-route extrapolations are required.
考量吸收与生物利用度间的潜在差异,尤其在需要进行给药途径间外推时
Bioaccumulation potential should be considered.
应考量生物蓄积潜力
Systemic Toxicity
全身毒性
Summarize relevant acute, subacute/subchronic and chronic toxicity studies.
总结相关的急性、亚急性 / 亚慢性和慢性毒性研究
Indicate relevance of data to humans.
说明数据对人类的相关性
Identify critical study (or studies) for evaluating human systemic toxicity potential.
确定用于评估人体全身毒性潜力的关键研究(一项或多项)
Sensitization Potential/Local Irritation
致敏潜力 / 局部刺激性
Relevant available clinical and non-clinical data (supplemented with in silico evaluation, if justified) should be summarized.
应总结现有相关临床和非临床数据(若合理,可辅以计算机模拟评估)
Regulatory classifications (or lack thereof) may be leveraged as pertinent.
可合理利用监管分类(或无相关分类的情况)
Developmental and Reproductive Toxicity (DART)
发育与生殖毒性(DART)
In addition to summarizing available DART studies, data and/or classifications with respect to endocrine disrupting properties should evaluated and included.
除总结现有 DART 研究外,还应评估并纳入与内分泌干扰特性相关的数据和 / 或分类
Genotoxicity and Carcinogenicity
遗传毒性与致癌性
Summarize available data and indicate potential relevance to humans.
总结现有数据并说明对人类的潜在相关性
If data are not available, in silico methods consistent with ICH M7 should be used for evaluation (Note: ICH M7 Class 4 is not applicable to leachables).
若无数据,应使用符合 ICH M7 的计算机模拟方法进行评估(注:ICH M7 的 4 类不适用于可浸出物 )
Mechanism(s) for genotoxicity and/or carcinogenicity should be provided if applicable as this is particularly pertinent for acceptable exposure determinations.
若适用,应提供遗传毒性和 / 或致癌性的作用机制,因这对确定可接受暴露量尤为关键
Additional Information
补充信息
Additional pertinent information to the safety assessment should also be included as available.
应纳入现有与安全评估相关的补充信息(如有)
Examples: Existing heath-based risk limit/assessments, clinical and epidemiological data, toxicological data from similar/related compounds
示例:现有基于健康的风险限值 / 评估、临床和流行病学数据、来自类似 / 相关化合物的毒理学数据
Acceptable Exposure Calculations
可接受暴露量计算
The PDE concept has been implemented as a health-based exposure limit in ICH guidelines in addition to other health-based limits such as the Acceptable Intake (AI).
在 ICH 指南中,除可接受摄入量(AI)等其他基于健康的限值外,允许日暴露量(PDE)概念也被用作基于健康的暴露限值。
The process for calculation of a PDE is generally aligned across these guidelines.
这些指南中 PDE 的计算流程基本一致。
This same basic approach has been used to generate PDE values in support of the identified qualification thresholds of the current guideline (with the inclusion of additional modifying factors for bioavailability and for when a read-across approach is used).
本指南中,为支持已确定的资质认定阈值,也采用了相同的基本方法来生成 PDE 值(包括针对生物利用度和使用关联方法时的额外修正因子)。
This approach is briefly described and summarized below and may be used as the basis for an acceptable exposure level for a leachable in a specific drug product.
下文简要描述和总结了该方法,其可作为特定药品中可浸出物可接受暴露量的基础。
Although the method for deriving an acceptable exposure level described here is based on the PDE methodology described in other ICH guidelines, it should be noted that the acceptable exposure may not necessarily be the same as the PDE.
尽管此处描述的可接受暴露量推导方法基于其他 ICH 指南中所述的 PDE 方法,但需注意,可接受暴露量未必与 PDE 完全一致。
Whereas the PDE is by definition an exposure level for lifetime and is applicable across many products, the product-specific acceptable exposure takes into account the duration of exposure and maximum daily dose.
PDE 按定义是终身暴露水平,适用于多种产品,而特定产品的可接受暴露量则需考虑暴露持续时间和最大日剂量。
Subsequent to review and evaluation of the available data and information for the leachable as described above, the derivation process begins with the selection of an appropriate point of departure (PoD) and then applying modifying factors (F1–F7).
在按上述内容审查和评估可浸出物的现有数据和信息后,推导过程首先是选择合适的起始点(PoD),然后应用修正因子(F1–F7)。
The most relevant study should be used to select the PoD, taking into consideration the species used, the route and duration of exposure, the toxicological endpoints monitored, and the quality of the study data, if justified, it may not always be necessary to select the lowest NO(A)EL as a PoD.
应使用最相关的研究来选择 PoD,需考虑所用物种、暴露途径和持续时间、监测的毒理学终点以及研究数据的质量;若有合理依据,未必一定要选择最低的未观察到(有害)效应水平(NO (A) EL)作为 PoD。
Previous guidelines have used specific modifying factors for inter- and intraspecies variability (F1 and F2, respectively), duration of the study from which the PoD is taken (F3), severity of the toxicity (F4), and a factor to account for the absence of a NOAEL (F5).
先前的指南已使用特定修正因子来分别考虑种间和种内变异(分别为 F1 和 F2)、PoD 所来源研究的持续时间(F3)、毒性严重程度(F4)以及无未观察到不良反应水平(NOAEL)时的修正因子(F5)。
As leachables cover a wide chemical space, bioavailability via various administration routes may vary.
由于可浸出物涉及广泛的化学物质,其通过不同给药途径的生物利用度可能存在差异。
Since toxicity data are often only available for a single route, the incorporation of an additional modifying factor (F6) is recommended in the current guideline to account for differences in bioavailability when route-to-route extrapolation is required.
由于毒性数据通常仅针对单一途径,本指南建议加入额外修正因子(F6),以在需要进行给药途径间外推时考虑生物利用度的差异。
Additionally, as noted previously, a PoD from a surrogate compound (read across approach) may also sometimes be necessary.
此外,如前文所述,有时可能需要使用来自替代化合物(关联方法)的 PoD。
Thus, another modifying factor (F7) to account for uncertainty related to using this surrogate compound is recommended.
因此,建议使用另一个修正因子(F7)来考虑与使用该替代化合物相关的不确定性。
As the criteria for selecting values for F1–F5 have been detailed in existing guidelines, they are not repeated here.
由于 F1–F5 的取值标准已在现有指南中详细说明,此处不再赘述。
However, the newly introduced modifying factors (F6 and F7) pertinent to leachables are summarized below.
但下文总结了新引入的与可浸出物相关的修正因子(F6 和 F7)。
F6 = A variable factor to account for route of exposure extrapolation (e.g., oral to parenteral).
F6 = 用于考虑暴露途径外推的可变因子(例如,从口服到肠外)。
In the absence of sufficient toxicity data on the leachable via the intended route of exposure of the drug product, F6 should be used to adjust for any pertinent difference in bioavailability between the PoD study route of administration and the drug product route of exposure.
若缺乏可浸出物通过药品预期暴露途径的充分毒性数据,应使用 F6 来调整 PoD 所来源研究的给药途径与药品暴露途径之间在生物利用度上的任何相关差异。
Ideally, F6 should be based on bioavailability of the parent compound.
理想情况下,F6 应基于母体化合物的生物利用度。
If a radiolabel study is used, it should be referred to as absorption because it is not clear if the radiolabel is the parent, a metabolite, or a combination of parent and metabolites.
若使用放射性标记研究,应称为吸收度,因为无法明确放射性标记是母体化合物、代谢物,还是母体与代谢物的混合物。
If the quality of data is good, the relative bioavailability estimate can be used to directly inform F6.
若数据质量良好,相对生物利用度估算值可直接用于确定 F6。
When there is significant uncertainty for the bioavailability estimate, default factors may alternatively be applied.
当生物利用度估算存在显著不确定性时,可改用默认因子。
For example, when using oral toxicity data to derive a parenteral acceptable exposure level:
例如,当使用口服毒性数据推导肠外可接受暴露量时:
F6= 100 when oral bioavailability is <1% (divide by a modifying factor of 100)
当口服生物利用度 < 1% 时,F6=100(除以修正因子 100)
F6= 10 when oral bioavailability is ≥ 1% and <50% (divide by a modifying factor of 10)
当口服生物利用度≥1% 且 < 50% 时,F6=10(除以修正因子 10)
F6= 2 when oral bioavailability is ≥50% and <90% (divide by a modifying factor of 2), and
当口服生物利用度≥50% 且 < 90% 时,F6=2(除以修正因子 2),以及
F6=1 when oral bioavailability is ≥ 90% (divide by a modifying factor of 1)
当口服生物利用度≥90% 时,F6=1(除以修正因子 1)
In the absence of sufficient in vivo data, additional approaches should be employed as part of a weight-of-evidence strategy or in lieu of in vivo data.
在缺乏充足体内数据时,应采用额外方法,作为证据权重策略的一部分,或替代体内数据。
For example, a NAM approach (combining in vitro data estimating absorption and internal clearance, with an in silico PBPK model) can be used to generate data to assess bioavailability if properly supported and scientifically justified.
例如,若有充分支持且科学合理,非动物实验方法(NAM,结合估算吸收和内在清除率的体外数据与计算机模拟的生理药代动力学(PBPK)模型 )可用于生成数据以评估生物利用度。
Alternatively, a default modifying factor of 100 is suggested for F6, with smaller values requiring justification (e.g., reasoning based on the physicochemical characteristics of the compound).
或者,建议为 F6 设定默认修正因子 100,使用更小数值需进行论证(如基于化合物理化特性的推理 )。
When suitable bioavailability data are available for a surrogate molecule allowing a read-across approach these data may be leveraged to inform the bioavailability estimate, if sufficiently justified.
若有适用于替代分子的生物利用度数据且可采用关联方法,在充分论证后,这些数据可用于辅助生物利用度估算。
For some routes, such as inhalation, additional considerations are warranted when determining an appropriate F6 value.
对于某些给药途径(如吸入),确定合适的 F6 值时需额外考量。
For example, for an inhalation toxicology study, data on respiratory tract deposition, respiratory absorption rate and pulmonary metabolism may inform on F6.
例如,对于吸入毒理学研究,呼吸道沉积、呼吸吸收率和肺部代谢的数据可能会为 F6 提供参考。
For dermal routes, if toxicokinetic data are available these can be used to estimate the systemic dose.
对于经皮给药途径,若有毒代动力学数据,可用于估算全身剂量。
The parenteral QT can be referred to when evaluating the estimated total daily systemic dose of the leachable.
评估可浸出物的每日估算全身总剂量时,可参考肠外定量阈值(QT)。
In the absence of toxicokinetic data, when extrapolating from dermal dose to systemic dose, a default absorption of 70% or 50% is assumed to be sufficiently conservative for most organic solvent-based dilutes and water-based or dispersed dilutes, respectively.
缺乏毒代动力学数据时,从经皮剂量外推至全身剂量,对于大多数基于有机溶剂的稀释液和水基或分散稀释液,默认分别采用 70% 或 50% 的吸收率,被认为具有足够保守性。
If both the molecular weight is greater than 500 and the logPow is either below –1 or above 4, a default absorption factor of 10% is assumed.
若分子量大于 500 且辛醇 - 水分配系数对数值(logPow )小于 - 1 或大于 4,默认采用 10% 的吸收因子。
Leachables may penetrate the skin to a greater extent when present in dermal drug products that are formulated for enhanced percutaneous absorption or where skin integrity may be compromised.
当可浸出物存在于为增强经皮吸收而配制的皮肤用药品中,或皮肤完整性可能受损时,其渗透皮肤的程度可能更高。
A higher rate of absorption should be assumed in such cases.
此类情况下,应假设更高的吸收率。
F7= A variable factor that may be applied if a Read Across Approach is used.
F7 = 采用关联方法(Read Across Approach )时可能应用的可变因子。
When read across strategy is utilized, a factor of up to 5 may be used depending on the level of (dis)similarity to the leachable compound of interest.
采用关联策略时,根据与目标可浸出化合物的(不)相似程度,最多可使用 5 倍的因子。
In general, when a surrogate is considered similar based on the criteria described in this guideline, an F7 of 1 may be applicable.
一般而言,若根据本指南所述标准,替代物被认为具有相似性,F7 可取 1。
References
参考文献
Copies of articles (or other documents) referenced to support a proposed PDE should be provided.
应提供用于支持拟议允许日暴露量(PDE)的参考文献(或其他文件 )副本。
Margin of Safety (MOS) and justification for leachable levels higher than a calculated acceptable exposure level or established PDE
安全边际(MOS)及可浸出物水平高于计算得出的可接受暴露水平或既定 PDE 的论证
For each substance for which an acceptable exposure level (e.g., PDE or AI) has been determined, a margin of safety can be calculated using the following formula:
对于每个已确定可接受暴露水平(如 PDE 或 AI )的物质,可使用以下公式计算安全边际:
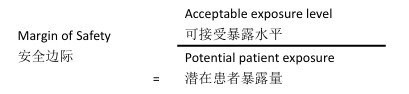
For any substances with an MOS <1, risk mitigation measures (such as the selection of alternate materials) that might reduce or eliminate the leachable of concern should be considered.
对于任何安全边际(MOS)<1 的物质,应考虑采取风险缓解措施(如选择替代材料 ),以减少或消除相关可浸出物。
Alternatively, it should be demonstrated that a limit greater than the acceptable exposure level (e.g., PDE) does not pose a safety concern for a specific drug product.
或者,应证明高于可接受暴露水平(如 PDE )的限值,对特定药品而言不会引发安全性问题。
An acceptable exposure level to a leachable higher than the calculated or established PDE may be acceptable in certain cases, taking into account relevant product-specific considerations. These cases could include, but are not limited to, the following situations:
在某些情况下,考虑到特定产品的相关因素,可浸出物高于计算得出或既定 PDE 的可接受暴露水平可能是可接受的。这些情况包括但不限于以下情形:
Intermittent administration of the drug to patients;
向患者间歇性给药;
Short term administration (i.e., 30 days or less);
短期给药(即 30 天或更短 );
Limited patient population (e.g., adult males only);
患者人群有限(如仅成年男性);
Specific indications (e.g., life-threatening, unmet medical needs, rare diseases).
特定适应症(如危及生命、未满足的医疗需求、罕见病)。
Additionally, it should be noted, that for drugs administered for less than lifetime to the patient, it may be appropriate to use a lower value for F3 than would usually be applied where a toxicity study of short-term exposure is selected as PoD.
此外,应注意,对于给药持续时间短于患者 lifetime(一生 )的药品,若选择短期暴露毒性研究作为PoD,使用比通常更低的 F3 值可能是合适的。
In this case an acceptable exposure level is derived, as opposed to PDE. If additional animal studies are available with longer duration, these may have NOAEL values based on findings that may not be relevant to shorter term exposures and therefore may not be the most appropriate PoD for a given drug product.
此时推导得出的是可接受暴露水平,而非 PDE。若有持续时间更长的其他动物研究,其未观察到不良反应水平(NOAEL )可能基于与短期暴露不相关的发现,因此对特定药品而言可能并非最恰当的 PoD。
However, while toxicity studies of short-term exposure may be acceptable as a PoD in this circumstance, this does not include LD50 studies.
不过,虽然短期暴露毒性研究在此情况下可作为 PoD,但不包括半数致死量(LD50 )研究。
In cases where a product is administered intermittently, a subfactor approach for F2 as described in ICH Q3D can be applied if supported by data. Alternatively, the value for F3 can be modified.
若产品为间歇性给药,如有数据支持,可采用 ICH Q3D 中所述的 F2 子因子方法。或者,可调整 F3 的值。
Table A.5.1: Example considerations for a weight of evidence justification when qualification of leachables is necessary. Non-animal methods should be prioritized where possible.
表 A.5.1:当需要对可浸出物进行资质认定时,证据权重论证的示例考量因素。应尽可能优先采用非动物实验方法。
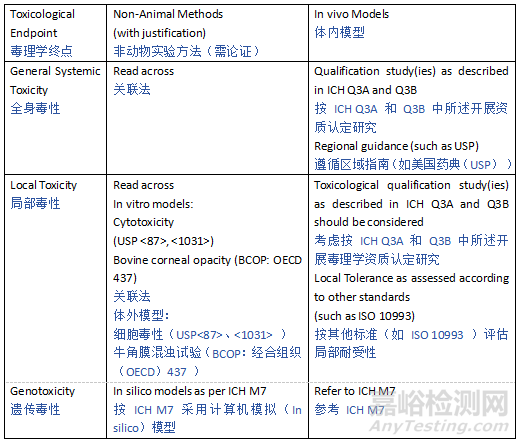
Appendix 6: Monographs for Class 1 Leachables
附录 6:1 类可浸出物各论
Benzo[a]pyrene
苯并 [a] 芘
Summary of Acute Acceptable Exposure Level and Chronic PDE Values for Benzo[a]pyrene (CAS# 50-32-8)
苯并 [a] 芘(CAS 编号 50 - 32 - 8)的急性可接受暴露水平及慢性允许日暴露量(PDE)值总结
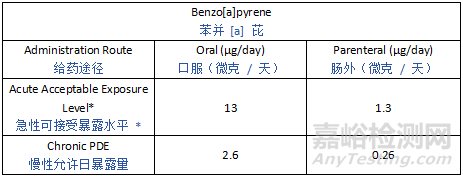
*Acute acceptable exposure level is applicable to ≤1 - month daily administration
* 急性可接受暴露水平适用于每日给药≤1 个月的情况
Introduction
介绍
Benzo[a]pyrene (BaP) is a polycyclic aromatic hydrocarbon (PAH) consisting of five fused benzene rings.
苯并 [a] 芘(BaP)是由五个稠合苯环组成的多环芳烃(PAH)。
It is not produced or used commercially but is formed as a result of incomplete combustion of organic matter.
它并非通过商业途径生产或使用,而是由有机物不完全燃烧产生。
BaP may leach from materials in which carbon black is present.
苯并 [a] 芘可能从含有炭黑的材料中浸出。
BaP is a mutagenic carcinogen and as such, control according to the current version of ICH M7 is appropriate, in addition to the relevant Acceptable Exposure or PDE values derived below.
苯并 [a] 芘是致突变致癌物,因此,除下文推导的相关可接受暴露或允许日暴露量(PDE)值外,按照现行版 ICH M7 进行控制是恰当的。
Based on a non - mutagenic endpoint, two oral and two parenteral values for BaP were developed for ICH Q3E.
基于非致突变终点,为 ICH Q3E 推导了苯并 [a] 芘的两个口服和两个肠外给药的数值。
Safety Summary and Limiting Non - Mutagenic Toxicity
安全性总结及限制性非致突变毒性
Oral exposure to BaP has been shown to result in developmental toxicity (including developmental neurotoxicity), reproductive toxicity, and immunotoxicity in repeat dose toxicity studies, including adult and juvenile animals.
在重复给药毒性研究(包括成年和幼年动物研究)中,口服暴露于苯并 [a] 芘已被证明会导致发育毒性(包括发育神经毒性)、生殖毒性和免疫毒性。
Overall, human studies report toxicological effects that are generally analogous to those observed in animals, and provide qualitative, supportive evidence for hazards associated with BaP exposure.
总体而言,人体研究报告的毒理学效应与在动物中观察到的效应大致相似,为苯并 [a] 芘暴露相关危害提供了定性的支持性证据。
Based on critical non - mutagenic effects of BaP, the non - GLP oral developmental toxicity study in neonatal rat (Chen et al., 2012) was selected as the PoD study for oral and parenteral PDE derivation.
基于苯并 [a] 芘的关键非致突变效应,选择新生大鼠的非良好实验室规范(GLP)口服发育毒性研究(Chen 等人,2012 年)作为推导口服和肠外给药 PDE 的起始点(PoD)研究。
Oral Acceptable Exposure and PDE
口服可接受暴露量和 PDE
The rat neurodevelopmental study by Chen et al., 2012 administered doses of BaP at 0, 0.02 mg/kg, 0.2 mg/kg, and 2 mg/kg on postnatal day 5 to 11 by oral gavage.
Chen 等人 2012 年开展的大鼠神经发育研究中,在新生大鼠出生后第 5 天至第 11 天,通过灌胃给予苯并 [a] 芘,剂量分别为 0、0.02 mg/kg、0.2 mg/kg 和 2 mg/kg。
Altered responses in three behavioral tests (Morris water maze, elevated plus maze, and open field tests) were selected to represent the critical effect of abnormal behavior, due to the consistency of the observations across groups/studies (i.e., each of these responses were affected in two separate cohorts of rats, including testing as juveniles and as adults; similar effects in these behavioral tests were observed across studies) and sensitivity of these responses, and the observed dose - response relationship of effects across dose groups.
由于在不同组 / 研究中观察结果具有一致性(即这些反应在两组不同的大鼠中均受到影响,包括幼鼠和成年鼠测试;在不同研究中观察到这些行为测试有类似效应)、这些反应的敏感性以及在不同剂量组中观察到的效应剂量 - 反应关系,选择三项行为测试(Morris 水迷宫、高架十字迷宫和旷场试验)中的反应改变来代表异常行为的关键效应。
Benchmark dose (BMD) modeling for each of the three endpoints resulted in BMD lower bound for 1 standard deviation (BMDL1SD) values in the range 0.092−0.16 mg/kg - day.
对这三个终点分别进行基准剂量(BMD)建模,得出 1 倍标准偏差的基准剂量下限(BMDL1SD)值在 0.092 - 0.16 mg/kg・天范围内。
Taking the lower end of the range, 0.092 mg/kg - day, was selected to represent the PoD from the neurodevelopmental study.
选取该范围的下限 0.092 mg/kg・天,作为神经发育研究的PoD。
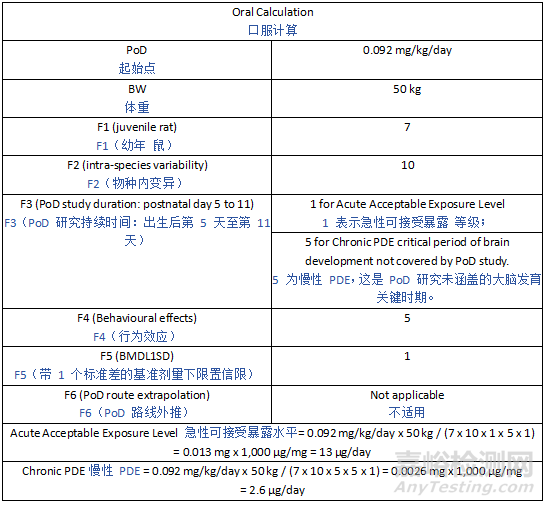
Parenteral Acceptable Exposure and PDE
肠外可接受暴露量和PDE
In the absence of parenteral administration repeat dose toxicity studies, the same POD study was used to derive the parenteral PDE with the inclusion of a bioavailability modifying factor (F6), based on physiochemical characteristics of BaP (MW = 252.3 g/mol and predicted LogP 3.0 (PubChem, 2024)).
在没有进行肠外给药重复剂量毒性研究的情况下,使用相同的 POD 研究,根据BaP的物理化学特性(MW = 252.3 g/mol和预测LogP 3.0(PubChem,2024)),得出肠外 PDE,其中包括生物利用度修正因子(F6)。
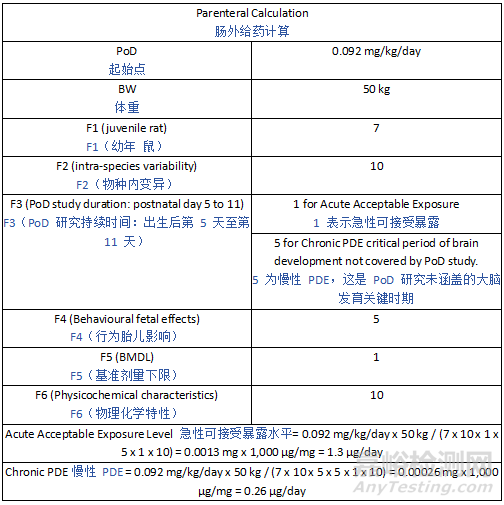
REFERENCES
参考文献
International Conference on Harmonisation (2023). M7(R2): Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk.
PubChem (2024) Compound Summary for CID 2336, Benzo[a]pyrene, National Center for Biotechnology Information. Retrieved May 2, 2024, from https://pubchem.ncbi.nlm.nih.gov/compound/Benzo_a_pyrene.
Chen, C; Tang, Y; Jiang, X; Qi, Y; Cheng, S; Qiu, C; et al. (2012). Early postnatal benzo(a)pyrene exposure in Sprague-Dawley rats causes persistent neurobehavioral impairments that emerge postnatally and continue into adolescence and adulthood. Toxicol Sci 125: 248-261. https://academic.oup.com/toxsci/article/125/1/248/1668305.
Bisphenol A
双酚A
Summary of Acute Acceptable Exposures and Chronic PDE Values for Bisphenol A (CAS# 80-05-7)
双酚 A (CAS# 80-05-7 )急性可接受暴露量和慢性 PDE 值总结
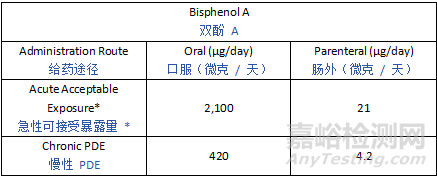
*Acute Acceptable Exposure value is applicable to ≤1-month daily administration
* 急性可接受暴露值适用于每日给药≤1 个月的情况
Introduction
介绍
Bisphenol A (BPA) is 4,4'-methanediyldiphenol where the methylene hydrogens are replaced by two methyl groups.
双酚 A(BPA)是 4,4'- 亚甲基二苯酚,其亚甲基上的氢被两个甲基取代。
It is a key building block of polycarbonate plastic and a precursor for the manufacturing of monomers of epoxy resins.
它是聚碳酸酯塑料的关键组成部分,也是环氧树脂单体生产的前体。
BPA may be present in primary packaging material and manufacturing equipment used in the manufacturing process of medicines, in medicine containers, medicine/device combinations, and in parenteral nutrition bags (Parris et al, 2020).
双酚 A 可能存在于药品生产过程中使用的初级包装材料和生产设备、药品容器、药械组合产品以及肠外营养袋中(Parris 等人,2020 年)。
Safety Summary and Limiting Toxicity
安全性总结及限制性毒性
BPA is not mutagenic and non-genotoxic.
双酚 A 无致突变性,也无遗传毒性。
ECHA listed BPA capable of producing skin sensitization responses in humans and may damage fertility or the unborn child.
欧洲化学品管理局(ECHA)将双酚 A 列为可能在人体中引起皮肤致敏反应,并可能损害生育能力或未出生胎儿的物质。
BPA is not a skin irritant; however, it is irritating to the eye (ECHA, 2024).
双酚 A 对皮肤无刺激性,但对眼睛有刺激性(ECHA,2024 年)。
The European Medicines Agency (EMA) obligates the use of an apical endpoint to minimize uncertainty in relation to human health risk assessment; ICH Q3E is aligned with EMA, and therefore non-mutagenic PDEs were derived for evaluation of BPA as a potential leachable in pharmaceutical products (EFSA EMA, 2023).
欧洲药品管理局(EMA)要求使用 apical 终点( apical endpoint,指整体生物学效应终点 )以最大限度减少人体健康风险评估中的不确定性;ICH Q3E 与 EMA 保持一致,因此推导了非致突变性允许日暴露量(PDE),用于评估双酚 A 作为药品中潜在可浸出物的情况(欧洲食品安全局与欧洲药品管理局,2023 年)。
Oral Acceptable Exposure and PDE
口服可接受暴露量和 PDE
BPA was tested in a two-generation study in mice (Tyl et al 2008).
在小鼠中开展了双酚 A 的两代研究(Tyl 等人,2008 年)。
The GLP and OECD 416-compliant study in mice, evaluated dietary BPA concentrations of 0, 0.018, 0.18, 1.8, 30, 300, or 3500 ppm (approximately 0.003, 0.03, 0.3, 5, 50, or 600 mg/kg/day) ad libitum.
这项符合良好实验室规范(GLP)和经合组织(OECD)416 号准则的小鼠研究,评估了饮食中双酚 A 浓度为 0、0.018、0.18、1.8、30、300 或 3500 ppm(约 0.003、0.03、0.3、5、50 或 600 mg/kg/ 天)时的自由摄食情况。
Concurrent positive control group of dietary 17β-estradiol (0.5 ppm; 28 per sex) was included to evaluate potential for endocrine disruption.
同时纳入饮食中含 17β- 雌二醇(0.5 ppm;雌雄各 28 只)的阳性对照组,以评估内分泌干扰潜力。
F0 generation animals received respective formulations in the diet for 8 weeks prior to mating (i.e., until ~14 weeks of age).
F0 代动物在交配前 8 周(即直至约 14 周龄)的饮食中接受相应制剂。
The animals were then mated for a period of 14 days.
随后动物交配 14 天。
Animals continued dosing through gestation (~20 days) and lactation (3 weeks).
动物在妊娠期(约 20 天)和哺乳期(3 周)持续给药。
No BPA-related effects at any dose were observed for adult mating, fertility or gestational indices, ovarian primordial follicle counts, estrous cyclicity, pre-coital interval, offspring sex ratios or post-natal survival, sperm parameters or reproductive organ weights or histopathology (including the testes and prostate).
在任何剂量下,均未观察到与双酚 A 相关的对成年动物交配、生育力或妊娠指数、卵巢原始卵泡计数、发情周期、交配前间隔、后代性别比例或出生后存活率、精子参数或生殖器官重量及组织病理学(包括睾丸和前列腺)的影响。
Systemic effects observed in adults were centrilobular hepatocyte hypertrophy at ≥300 ppm, reduced body weight, increased kidney and liver weights, centrilobular hepatocyte hypertrophy, and renal nephropathy in males.
在成年动物中观察到的全身效应为:≥300 ppm 剂量组出现小叶中心肝细胞肥大;雄性动物出现体重下降、肾和肝重量增加、小叶中心肝细胞肥大以及肾肾病。
In conclusion, the NOAEL for reproductive toxicity was 300 ppm (~50 mg/kg/day) and NOEL for adult (F0) systemic toxicity was 30 ppm (~5 mg/kg/day).
结论:生殖毒性的未观察到不良反应水平(NOAEL)为 300 ppm(约 50 mg/kg/ 天),成年(F0 代)全身毒性的未观察到效应水平(NOEL)为 30 ppm(约 5 mg/kg/ 天)。
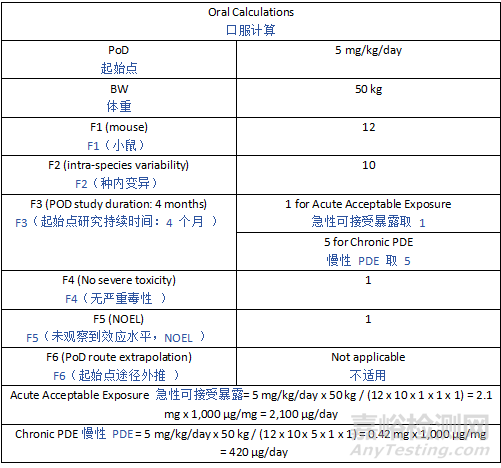
Parenteral Acceptable Exposure and PDE
肠外可接受暴露量和允许日暴露量(PDE)
In the absence of parenteral administration repeat dose toxicity studies, the same POD study was used to derive the parenteral PDE with the inclusion of a bioavailability modifying factor (F6).
在缺乏肠外给药重复剂量毒性研究的情况下,采用相同的起始点(POD)研究,并纳入生物利用度修正因子(F6),来推导肠外给药的 PDE。
Oral systemic bioavailability of unconjugated BPA of 2.8% in rats and less than 1% in mice, monkey and dogs was reported (ANSES, 2013).
有报道称,非结合型双酚 A(BPA)在大鼠中的口服全身生物利用度为 2.8%,在小鼠、猴和犬中小于 1%(法国国家食品、环境及劳动卫生署(ANSES),2013 年)。
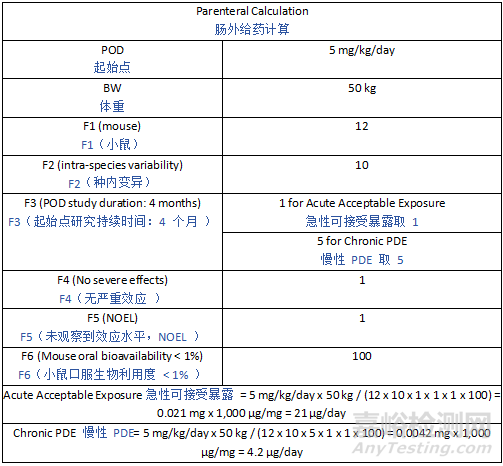

来源:GMP办公室