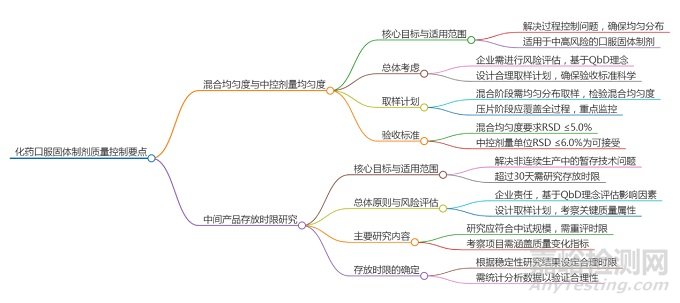
在化药口服固体制剂(如片剂、胶囊剂、颗粒剂等)的生产中,确保活性成分在单位剂量中的均匀分布以及中间产品在暂存期间的稳定性,是保障最终成品质量、安全性和有效性的核心环节。为贯彻ICH提出的“质量源于设计”(QbD)理念,提升生产过程中的风险控制水平,CDE发布了《化药口服固体制剂混合均匀度和中控剂量单位均匀度研究技术指导原则(试行)》和《化药口服固体制剂中间产品存放时限研究技术指导原则》。这两份文件为制药企业提供了明确的技术框架和实施策略。
01混合均匀度与中控剂量单位均匀度研究:确保含量均匀的基石
混合(尤其是最终混合)和压片/填充步骤是口服固体制剂生产的关键工艺步骤。混合均匀度(指混合物料的均匀性)和中控剂量单位均匀度(指未经包衣/包装的单片或单粒胶囊的含量均匀性)直接决定了成品含量均匀度能否达标。
1.核心目标与适用范围:
a)旨在提供一种研究策略,解决工业上关注的过程控制问题,确保每个单位剂量含有均等的活性物质。
b)适用于需进行含量均匀度测定的口服固体制剂(中国药典要求),特别是经风险评估认为混合或压片/填充步骤存在中高风险的品种。应对产品全生命周期中的关键批次(如工艺验证、临床研究、上市后变更批次)进行考察。
2.总体考虑:
a)制剂申请人/生产企业是责任主体,需基于QbD理念和产品/工艺特点进行风险评估。影响混合不均的因素包括物料性质(粒径分布、引湿性等)和工艺特点(湿法制粒、粉末直压等)。
b)需设计合理的取样计划,建立科学的验收标准,以充分反映物料混合效果并确保成品均匀性。
3.取样计划:
推荐分层取样,在预定时间/位置间隔收集样品,或在压片/填充过程中收集高风险位点(可能造成成品不均匀)的样品,监控最易引起成品差异的过程。
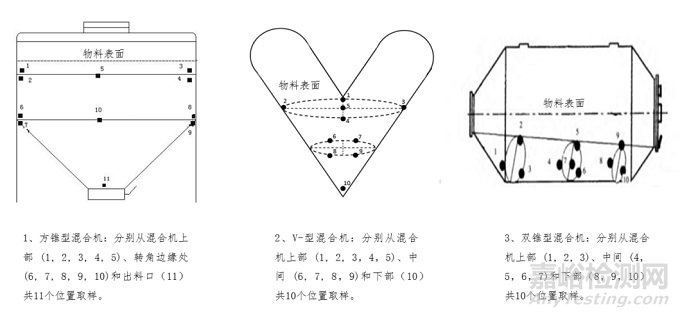
a)混合阶段取样:压片/填充前的最终混合物均匀度是基础,也是取样研究的最佳阶段,原则上不应跳过此阶段研究。取样点应均匀分布且有代表性,需结合设备结构确定死角,涵盖全部物料及最易混合不均的位置(如滚筒式混合机的上、中、下层及卸料区,具体位置参考示意图1)。至少在混合设备/中间体容器中选10个取样点,每点取至少3份样品。单份样品量通常在1-10倍单位剂量范围内,应全量检测避免二次取样。若>3倍单位剂量需论证合理性。对多批次进行广泛取样,用统计方法评估混合均匀度,分析偏差原因(混合不充分、取样误差、物料聚集或多个因素的组合效应)。
b)压片/填充阶段取样:取样点必须覆盖全过程, 预设取样位置和间隔,覆盖整个运行过程,重点关注重要事件(如加料过程、停机再启动过程等)。整个批次中一般不少于20个取样点(包括开始和结束点),每点取样至少7个剂量单位。批量小、时长短等特殊情况可减少,但需充分科学说明。 对比混合物料均匀度与中控剂量单位数据,分析差异根本原因,考虑优化处方或工艺。
▲图1-取样示意图
4.验收标准(示例):
a)混合均匀度验收标准:
1)取至少10点,每点至少3份样品。
2)每点检测1份样,计算所有样品的RSD (n≥10),所有单值在均值±10.0%(绝对)内。
● RSD ≤ 5.0%:进行中控剂量单位均匀度测定。
● RSD > 5.0%:检测剩余样品(每点未检样品)混合均匀度。
3)测剩余样混合均匀度,计算所有样RSD (n≥30),所有单值在均值±10.0%(绝对)内。
● RSD ≤ 5.0%:进行中控剂量单位均匀度测定(至少20点,每点至少7个剂量单位)。
● RSD > 5.0%:调查变异性来源(产品/工艺问题 or 取样/检测误差)。若属取样/检测误差,进行中控剂量单位均匀度测定(至少20点,每点至少7个剂量单位)。若属产品/工艺原因,混合均匀性不可接受。
b)中控剂量单位均匀度验收标准:
1)至少20点在线取样(含开始和结束点,数值需重量校正),每点至少7个剂量单位。
2)测每点至少3单位,计算所有样RSD (n≥60),每点平均值在目标剂量90.0%-110.0%内,所有单值在75.0%-125.0%内。
● RSD ≤ 6.0%:可接受。
● RSD > 6.0%:检测剩余样品(每点所有未检剂量单位)。
3)测剩余样中控剂量单位均匀度,计算所有样RSD (n≥140),每点平均值在目标剂量90.0%-110.0%内,所有单值在75.0%-125.0%内。
● RSD ≤ 6.0%:可接受。
● RSD > 6.0%:批次不均匀。分析两个阶段数据,确定变异来源,改进工艺。
5.其他考虑:
a)推荐的取样计划和标准非绝对,遇特殊情况(取样点不足/需增加、取样量超出范围)可调整,但需提供充分的科学依据和论证,确保目标达成。
b)对于窄治疗指数或高活性药物,建议基于科学认知和风险评估,制定更严格的取样计划和验收标准。
02中间产品存放时限研究:保障暂存期间质量稳定的关键
口服固体制剂生产常涉及多个工序,中间产品(如处理后的原料药、黏合剂、总混颗粒、片芯、包衣液、包衣片)可能因生产安排需要暂存。存放时限指其在特定条件下存放并能维持符合既定质量标准的时间长度。
1.核心目标与适用范围:
1)旨在解决非连续生产中中间产品短暂存贮的技术问题,提供存放时限研究指导。
2)适用于化药口服固体制剂。当整个生产过程超过30天时,必须进行中间产品存放时限研究;不超过30天时,需通过风险评估决定是否进行。
2.总体原则与风险评估:
1)药品注册申请人/生产企业是责任主体。
2)应基于QbD理念和产品/工艺特点,对影响中间产品质量的因素(包装形式、存放温湿度等)进行风险评估。
3)设计合理取样计划,选择能反映质量变化、影响后续工艺及成品关键质量属性(CQAs)的项目作为考察指标。
3.主要研究内容:
1)样品要求:
● 研究应至少在中试规模批次进行。若研究批量未达到商业化规模,需承诺在商业化规模(含拟定场地)确认。
● 当处方、工艺、设备、贮藏条件、包材变更时,需通过风险评估判定是否需重新研究或确认时限。
● 至少选择一批中间产品确定时限。可基于物料特性等通过风险评估确定适当批次。多规格制剂可选代表性规格研究。
2)研究对象、考察时间与项目:
● 确定对象: 分析生产过程,根据需特殊存贮的环节、生产时间、环境与存贮条件的潜在影响确定研究对象(如原料药处理后、总混颗粒、片芯、包衣液、包衣片)。
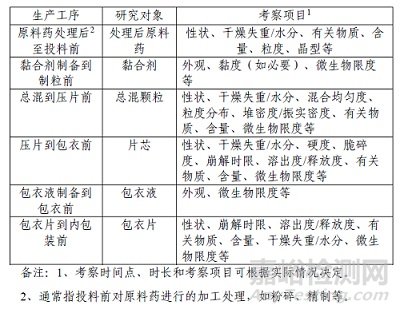
● 考察项目: 应能反映中间产品质量在放置过程中的变化(易变且影响成品质量、安全/有效性的项目),涵盖物理、化学、微生物特性(如性状、水分、含量、有关物质、粒度、均匀度、硬度、脆碎度、崩解、溶出、微生物限度等)。具体项目因剂型和工序而异(口服包衣片剂考察项目见示例表1)。
● 考察时间点与时长: 持续时间应涵盖拟定的最长存放时间。时间点至少包括开始、中间和结束时间,需科学设定。
▲表1-口服包衣片剂存放时限生产工序、研究对象、考察项目示例表
3)存放条件:
● 不应对后续工艺及中间产品质量、安全/有效性产生不良影响。
● 环境条件应与暂存区域相当,否则需说明合理性。
● 暂存容器应与商业化生产所用相同。若考察中必须减小容器尺寸,需使用材质相同、密闭系统等同的容器,并论证合理性。
● 对稳定性风险高(如易氧化)产品,关注容器顶部空间的潜在影响。存放时限研究应考虑最差情形(顶部空间与容器容量比例至少等于常规生产中可能的最大情形,特别是未装满容器)。
● 存在跨场地转移时,需考虑转移时间和条件,必要时进行加速试验证明短期偏离的稳定性。
4)质量标准与分析方法:
● 根据产品特点和质量控制需要合理设定研究用质量标准。
● 分析方法需经过方法学验证并能满足研究要求。
5)存放时限的确定:
● 根据稳定性研究结果和实际生产需要拟定合理时限。
● 必要时对数据进行统计学分析(变化趋势),判定各项目拟定限度与存放时限设定的合理性。
● 若数据显示存放期间含量降低、降解产物增加等,需考虑:1)对经最长存放时限研究的中间产品制成的成品进行加速/长期稳定性试验。2)缩短中间产品的存放时限,确保制剂成品合格。
03结论
《化药口服固体制剂混合均匀度和中控剂量单位均匀度研究技术指导原则(试行)》与《化药口服固体制剂中间产品存放时限研究技术指导原则》为制药企业构建了一套科学、系统的质控框架。前者聚焦于生产过程中活性成分分布的实时监控与保障,通过分层取样和严格的RSD标准确保每个药片/胶囊的剂量精准;后者则关注生产中断时中间产品质量的稳定维持,通过模拟实际暂存条件和多指标动态监测为安全存贮期提供数据支撑。两者相辅相成,共同作用于口服固体制剂生产链条的关键节点,确保持续稳定地生产出符合预定质量标准、安全有效的化药口服固体制剂,最终保障患者的用药安全与疗效。
参考文献
[1] 《化药口服固体制剂混合均匀度和中控剂量单位均匀度研究技术指导原则(试行)》,2022年01月.
[2] 《化药口服固体制剂中间产品存放时限研究技术指导原则》,2025年01月.
来源:注册圈
关键词:
化药口服固体制剂
均匀度