1.1 何时可选择同品种临床评价路径?
可以从以下几点综合考虑:
①申报产品基于现有成熟技术并预期用于该技术的成熟应用;
②申报产品已有前代产品或同系列产品通过注册临时试验上市,未出现可能引发重大风险或显著影响有效性问题;
③境内已有已上市的同品种产品;
④查阅审评发布的推荐临床评价路径清单;
⑤获取同品种产品的技术资料的可行性;
⑥临床数据的支持力度,目前同品种临床评价路径的思路是进行临床支持的数据必须涉及已比对的同品种器械,所以在选择此评价路径时候,前期需初步评价下比对的同品种器械其临床数据的数量和质量,若无足够的支持力度,需考虑更换其他同类产品或增加其他同类比对产品。
1.2 提交同品种器械的临床数据理由?
①法规要求通过对同品种器械临床或者临床使用获得的数据进行分析评价,证明申报产品的安全性、临床性能和/或有效性;
②确认同品种器械的安全有效性在现有认知性,是否已得到临床公认,风险收益是否在可接受范围内;
③充分识别同品种器械的临床有效性和使用风险,为申报产品的风险收益分析提供信息;
④充分识别同品种器械的临床风险,为风险管理(最小化临床风险)提供信息。
1.3 如何理解选择多个器械作为同品种器械开展临床评价?
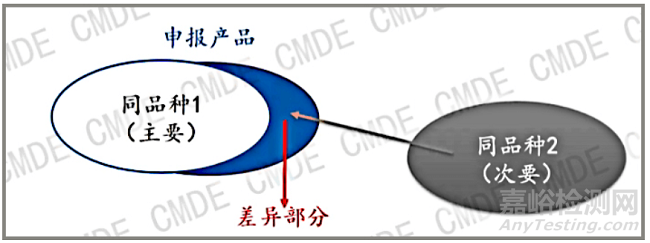
图 1 图片来源 CMDE
如图 1所示,当申报产品和同品种器械存在差异的时候,此时可以引入第二个同品种器械即“同品种2”作为差异性的证据,同品种器械1和2共同论证申报产品的安全有效性。此时需注意,不同的设计特征/适用范围在申报产品中组合时不应相互影响。如果存在互相影响,不能证据叠加。
1.4 某企业有个医疗器械品种和**上市产品一样,但是拿不到官方授权,能不能用同品种评价?
这个问题可以从以下几个方面来考虑:
①使用公开发表的数据,如公开发表的临床文献、临床数据以及官方网站公开的信息等,则不需要取得授权;
②拟使用同品种器械非公开数据和信息,比如产品性能、功能及其他关键技术特征,基于合法数据要求的基础上,注册申请人需要提供数据使用授权书,以保证数据来源的合法性。
③如果拿不到企业授权,申请人也可向国家局申请资料公开。
1.5 在中国境内的注册证过期了的产品,能不能用作同品种器械?
答案是:可以的。前提是该器械已在中国境内注册上市,并且没有由于该器械的安全性发生召回而被取消了注册证;其次,同品种器械并未停止生产或使用,同时尽可能选择注册证在有效期内的产品作为同品种器械。
1.6 某产品从II类升到III类,类别调整后的产品可采用自身临床数据作为医疗器械同品种比对临床数据进行临床评价吗?
答案是:可以的。但应充分收集其作为II类产品时的上市前和上市后数据,进行合理地总结和分析。主要关注申报产品是否在正常使用条件下,产品可达到预期性能;与预期受益相比较,产品的风险是否可接受;产品的临床性能和安全性是否均有适当的证据支持。
2、重点概念定义及相关解释
1) 对比器械
指的是注册申请人选择的,旨在将其临床数据用于支持申报产品临床评价的医疗器械。若对比器械与申报产品通过等同性论证,证明二者基本等同,则对比器械此时被认为是等同器械。
需从适用范围、技术特征、生物学特性等相关方面考虑对比器械的信息是否可用于申报产品的临床评价。
2)同品种医疗器械
当对比器械的适用范围、技术和/或生物学特性与申报产品具有广泛相似性时,可将其视为同品种医疗器械。同品种医疗器械包括可比器械和等同器械两种情形。
a. 等同器械
申报产品的适用范围与同品种医疗器械相同,技术特征和/或生物学特性与同品种医疗器械的相似程度使二者的安全性、临床性能和/或有效性不存在显著的临床差异,认为二者具有等同性。可通过等同器械的临床数据进行临床评估。
当申报产品的技术特征和生物学特征的对比器械存在差异时,需提交充分的科学证据证明二者的安全有效性,从而论证其等同性。
b. 可比器械
将申报产品与对比器械进行对比,虽然不能论证二者具有等同性,但二者在适用范围、技术特征和/或生物学特性具有广泛相似性时,可将对比器械视为可比器械,使用可比器械的临床数据用于支持申报产品的部分临床评价,作为申报产品临床证据的一部分。
3) 等同性论证
是指将适用范围相同的申报产品与对比器械在技术特征和生物学特性方面进行比对,证明二者基本等同的过程。基本等同包括两种情形:
(一)申报产品与对比器械具有相同的适用范围、技术特征和生物学特性;
(二)申报产品与对比器械具有相同的适用范围,相似的技术特征和生物学特性;有充分的科学证据证明申报产品与对比器械具有相同的安全有效性。
4) 临床证据
与医疗器械相关的临床数据及其评价。与其他设计验证及确认文件、产品描述、说明书和标签、风险分析及生产信息共同论证产品对安全和性能基本原则的符合性。
临床证据可用于支持产品上市,包括产品的适用范围以及对于产品安全性、临床性能和/或有效性的宣称。
5) 临床评价
采用科学合理的方法对临床数据进行分析评价,以确认医疗器械在其适用范围下的安全性、临床性能和/或有效性的持续进行的活动。临床评价由注册申请人实施,用于论证产品对安全和性能基本原则的符合性。
临床评价的结果是临床评价报告,可提供给监管部门进行审评。临床评价报告对临床数据及其质量进行详细阐述,论证临床数据如何证明产品对安全和性能基本原则的符合性。临床评价的输入主要是来源于临床试验报告、临床文献和临床经验的临床数据。
6) 临床数据
在医疗器械临床使用过程中产生的安全性、临床性能和/或有效性信息。
临床数据的来源包括:①申报产品上市前和上市后临床试验数据;②同品种医疗器械上市前和上市后的临床试验数据;③已发表和/或未发表的申报产品或同品种医疗器械的临床经验数据;④其他来源的临床经验数据,如登记研究、不良事件数据库和病历数据等。
7) 临床试验
为评价医疗器械的安全性、临床性能和/或有效性,在一例或多例受试者中开展的系统性的试验或研究(需注意在临床评价体系的临床试验的概念是大于《医疗器械临床试验质量管理规范》对于临床试验的概念)。
临床试验包括可行性试验、为获得上市批准而进行的试验,以及在上市批准后开展的试验。

来源:医学中心 ll.T
关键词:
医疗器械
临床评价