镓、锗、铟作为典型的稀散金属,很少有独立矿床存在。镓主要伴生在铝土矿中,少量存在于锡矿、钨矿、铅锌矿中。锗多伴生在含硫化物的铅、锌、铜等矿物中或以含锗褐煤存在。铟主要富集于硫化矿中,特别是闪锌矿中,还有一部分伴生在方铅矿、氧化铅矿等矿石中。随着镓、锗、铟需求量的增加,其战略地位越来越突出,国家“十四五”科技创新重点研发计划已经将镓、锗、铟等稀散金属的开发利用列为重点专项。在冶炼锌精矿时,镓、锗、铟会随着硫化锌矿石一起被浮选进入锌精矿中,因此建立灵敏度高、分析快速、结果可靠的测定稀散金属元素镓、锗、铟含量的方法已成为锌精矿分析的重点。
目前,镓、锗、铟的常见测定方法有分光光度法、原子荧光光谱法、原子吸收光谱法等。但是,锌精矿中稀散金属元素含量比较低,上述方法需要进行分离、萃取、富集等过程,操作繁琐、工作效率低,导致检测结果的不确定度增加。电感耦合等离子体质谱法(ICP-MS)具有快速、灵敏度高、准确度好、检出限低、动态线性范围宽、可多元素同时测定等特点,在地质样品稀散金属元素检测中的应用日益增多,包括在精矿中的应用。在国家标准GB/T 8151系列分析方法中,锗测定方法规定的检测范围(质量分数)为0.0005%~0.10%,铟测定方法规定的检测范围(质量分数)为0.0020%~0.10%,无法检出锌精矿中低于0.0005%的锗和低于0.0020%的铟,且缺乏镓的测定方法。一些锌精矿中镓质量分数最高可达0.03%,具有较高的回收价值。因此,建立锌精矿中镓、锗、铟同时测定的方法,对指导锌精矿冶炼生产和回收利用等工作具有重要意义。
本文根据锌精矿特性,采用硝酸-氢氟酸-高氯酸溶样,同时滴加2滴25%(体积分数,下同)硫酸溶液,避免铁氧化物析出。在进行ICP-MS分析时,探讨了共存元素、试剂的干扰,并选择内标元素103Rh校正71Ga、74Ge,187Re校正115In,同时以在线/离线公式校正74Se对74Ge以及115Sn对115In的干扰;当实际样品中铜含量过高时,通过进一步稀释来降低内标校正偏差。方法简便、快捷,测定结果准确。
1、试验方法
将锌精矿样品过筛后置于烘箱中加热,冷却备用。分取0.10g置于烧杯中,用少量水润湿,加入硝酸盖上表面皿,置于电热板上,以不超过260℃低温加热至溶液体积为3~5mL,取下稍冷,加入氢氟酸、高氯酸和25%硫酸溶液,继续加热至白烟冒尽。如果消解液仍呈黑色,说明样品含碳量较高,需要再加入高氯酸除尽有机物。取下稍冷,用水吹洗表面皿及烧杯壁,加入4mL50%(体积分数)硝酸溶液,盖上表面皿,以不超过260℃低温加热溶解盐类。取下冷却,将上述溶液移入容量瓶中,用水稀释至刻度,摇匀后用滤纸进行干过滤,弃去初滤液,收集中段滤液,供ICP-MS分析。其中,对于待测元素质量分数为0.00005%~0.0100%且铜质量分数小于8%的样品,直接按照上述步骤测定;对于待测元素质量分数为0.00005% ~0.0100%且铜质量分数不小于8%的样品,需要将滤液用2%硝酸溶液稀释1倍后进行测定;对于待测元素质量分数为0.0100%~0.0500%的样品,需要用2%硝酸溶液稀释10倍后进行测定。同步进行空白试验。
测定115In时,应同时测定115Sn,用公式(1)(ρSn/ρIn≤100的样品,在线理论值校正)或公式 (2)(ρSn/ρIn>100的样品,离线质量浓度校正)校正锡对铟的干扰;测定74Ge时,应同时测定77Se,用公式(3)校正硒对锗的干扰。
结合各待测元素的标准曲线和上述公式对检测结果进行校正和计算,所得结果减去空白值即为实际样品溶液中待测元素的质量浓度。
2、结果与讨论
2.1 称样量的选择
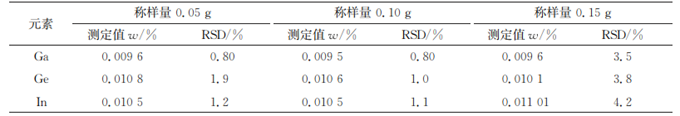
样品溶液中含有一定量的盐,这可能引起基体效应,导致检测结果出现偏差。虽然采用内标法可以校正盐引起的漂移,但是如果盐含量过大,内标校正的不确定度也同步增大。在满足分析灵敏度的前提下,可以通过减少称样量的方法降低样品溶液中盐含量,但是称样量也不能过少,否则检测结果易受仪器波动、试剂纯度、器皿洁净度等因素的影响。因此,试验比较了称样量分别为0.05,0.10,0.15g时,20次连续测定所得待测元素测定值以及测定值的相对标准偏差(RSD)的变化,结果见表1。
表1 称样量的选择(n=20)
由表1可知:称样量为0.05~0.15g时对待测元素测定值无显著影响;称样量为0.10g时,测定值的RSD较低,同时各待测元素以及内标的信号强度降低率较低(<3%)。因此,试验选择的称样量为0.10g。
2.2 溶样体系的选择
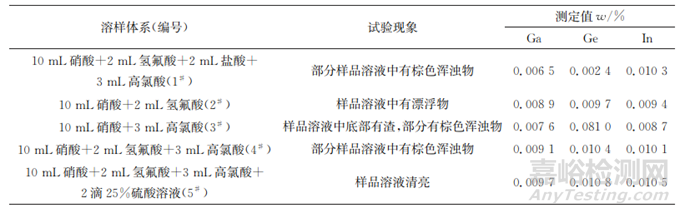
在处理实际样品时,既要保证样品能完全分解,又要避免溶样过程中待测元素挥发损失。溶样时如果引入盐酸,则会生成沸点分别为83,201℃的四氯化锗和三氯化镓,比较容易挥发,导致这两种元素测定结果偏低。当样品中这两种待测元素含量较低时,上述影响可能不显著。但是,一些锌精矿中锗质量分数高达0.05%,引入盐酸可能造成较大的锗损失,因此试验不能采用盐酸溶样且需避免有氯环境。镓、锗、铟的硝酸盐很稳定,不易挥发,因此试验可选择硝酸溶样。考虑到锌精矿中硫质量分数高达38%,用硝酸溶样易形成硫球,导致待测元素被包裹,测定准确度变差,故采用加入高氯酸加热至白烟冒尽的方法去除硫干扰。大部分锌精矿中还含有硅化合物,不能被硝酸-高氯酸体系溶解,因此还需在溶样体系中加入氢氟酸,以降低硅化合物夹杂的影响。约65%的锌精矿中铁质量分数大于8%,其中一些样品中铁的质量分数甚至高达20%,在溶样结束后,铁易形成不溶于硝酸的铁氧化物,在样品溶液中显示为棕色浑浊物,影响测定结果和仪器寿命,试验选择加入2滴25%硫酸溶液来防止铁氧化物的析出。为确定最优溶样体系,以一个含二氧化硅、硫 、铁质量分数分别为7.43%,33.5%,8.9%的样品为待测对象,分别按如下5种体系溶样,每种体系进行6次平行试验,结果见表2。
表2 溶样体系的选择
由表2可知:以体系1#溶样,镓、锗测定值较低,源于盐酸引起的待测元素挥发;以体系2#溶样,样品溶液中有漂浮物,3种待测元素测定值略低,源于生成的包裹性硫球夹杂;以体系3#溶样,样品溶液底部有渣,3种待测元素测定值偏低,源于样品中硅化合物夹杂;以体系4#溶样,样品溶液中产生了棕色浑浊物,源于生成的铁氧化物;在体系4#中补加2滴25%硫酸溶液,即以体系5#溶样时,样品溶液变清亮,3种待测元素测定值较高。因此,试验选择采用体系5#溶样。
2.3 待测元素同位素的选择
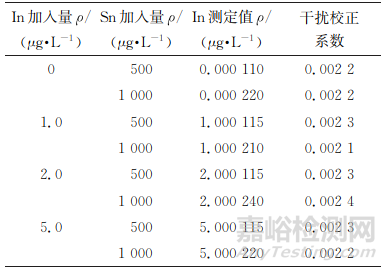
按照无干扰、丰度高的原则选择各待测元素同位素。镓可选的同位素有69Ga、71Ga,二者灵敏度接近,但考虑到138Ba2+丰度大(71.7%)且在锌精矿中质量分数高达2.5%,对69Ga存在较大干扰,因此试验选择71Ga为待测元素镓同位素。锗可选的同位素有72Ge、73Ge、74Ge、76Ge,其中74Ge的灵敏度较高,但是74Ge会受同量异位素74Se的干扰,考虑到锌精矿中硒质量分数普遍低于0.00005%,远低于锗,且74Se丰度低(0.9%),可通过校正公式(3)消除其干扰。铟可选的同位素有113In、115In,其中113In灵敏度低,受111Cd干扰大且不能通过公式校正消除;115In受同量异位素115Sn的干扰,但是锌精矿样品中锡质量分数通常低于 0.1%,且115Sn丰度小(0.36%),可按照公式(1)进行在线理论值校正得到准确结果。但是,针对个别锡质量分数较高(最高达0.3%)且铟含量较低(ρSn/ρIn>100)的样品,在线理论值校正可能会导致铟测定出现偏差。分别取铟标准溶液(100μg·L-1)0,1.00,2.00,5.00mL各2份于一组100mL容量瓶中,加入不同量锡标准溶液,按照仪器工作条件测定,结果见表3。
表3 115Sn对115In的干扰校正系数
由表3可知,115Sn对115In的干扰校正系数为0.0021~0.0024,以其平均值(0.0022)建立公式(2)离线校正ρSn/ρIn>100的样品中铟的测定值。
2.4 质谱干扰
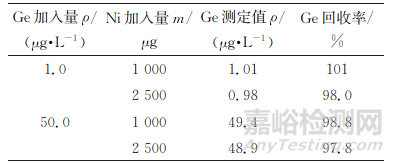
样品中的一些共存元素会形成多原子离子质谱干扰,因此试验考察了镍对74Ge的干扰。分别取1.00mL100μg·L-1锗标准溶液、5.00mL1000μg·L-1锗标准溶液各2份于一组100mL容量瓶中,加入不同量镍标准溶液,按照仪器工作条件测定,结果见表4。
表4 58Ni16O对74Ge的干扰试验
由表4可知:镍的存在对锗的准确测定干扰不大,可忽略。
一些质谱干扰为氧化物、多原子离子和同量异位素干扰等。对于镓、锗、铟,这些干扰主要来自氯、铈、钕、钼。氯会和等离子体中大量存在的氩、氧、氢结合,形成的71ArCl干扰71Ga,72Cl2干扰72Ge;钼形成的氧化物98Mo17O干扰115In;142Ce2+ 、142Nd2+干扰71Ga。其中:本试验不引入盐酸,避免了氯干扰;锌精矿中铈、钕、钼含量极微且干扰系数低,其干扰可以忽略不计。
2.5 非质谱干扰和内标的选择
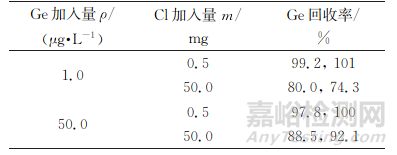
部分锌精矿中氯质量分数达0.5%,为考察氯对锗测定的影响,试验取1.00,50.00mL100μg·L-1混合标准溶液B各2份于聚四氟乙烯烧杯中,分别加入不同量氯(滴加盐酸),按照仪器工作条件测定,结果见表5。
表5 氯的干扰试验
由表5可知,当氯加入量为0.5mg时,锗回收率较理想,说明锌精矿氯质量分数高达0.5%时也不会引起锗测定偏差;当氯加入量为50mg (约2滴盐酸)时,锗测定结果明显降低,可能源于锗挥发损失,因此试验应避开含氯环境,同时不应引入含氯试剂。
分析某锌精矿企业近3年的检测数据可知,锌精矿中主量成分分布(质量分数中位值)为锌62%、铅10%、铁20% (约65%的样品中铁质量分数大于8%)、硫38% 、二氧化硅13%、钙10%、铜8% (约95%的样品中铜低于质量分数 2%)、镉1%、铝5%、镍 5%、钴 5%、锰 5%、钡 2.5%、硒<0.00005%,部分成分含量较高,可能影响待测元素的准确测定。试验选择采用内标法消除主量成分的干扰,内标元素同位素按照丰度高、无干扰,质量数与待测元素相近,样品中不含内标元素的原则来选择。锌精矿普查数据(中位值)显示,锌精矿中镧质量分数约0.0006%,钇质量分数约0.0005%,钪、铑、铱、铼的质量分数均小于 0.000001%,试验选择以45Sc、103Rh、187Re作内标元素,并考察了主要共存干扰元素铅、锌、铜对待测元素和内标元素的影响。在1.00,50.00μg·L-1混合标准溶液(内标元素的质量浓度为25.00μg·L-1)200mL中分别加入62mgZn、10mgPb、2mgCu、4mgCu、8mgCu以及62mgZn+10mgPb+4mgCu,按照仪器工作条件测定,结果见表6。
表6 主量元素干扰试验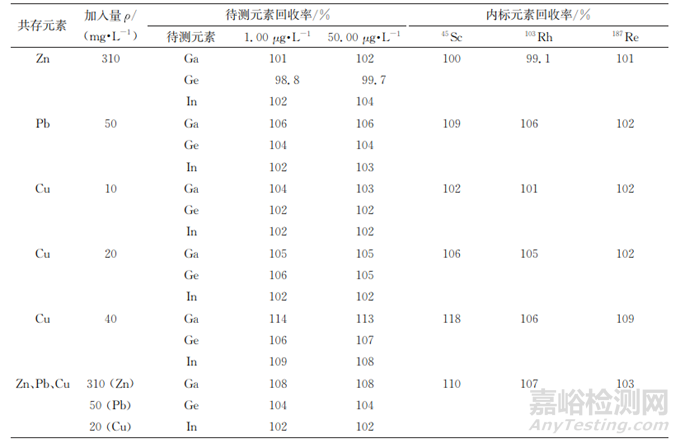
由表6可知:当存在50mg·L-1Pb、40mg·L-1Cu时,镓、锗、铟测定结果较高,其中镓测定结果偏高较多,其他共存元素干扰不大;钪回收率稍高于待测元素的,铑的回收率和镓、锗的基本一致,铼的回收率和铟的基本一致,因此试验选择以103Rh校正71Ga、74Ge、187Re校正115In。但是,当铜质量浓度不小于40mg·L-1(铜质量分数不小于8%)时,内标法校正结果有一定偏差,应进一步稀释样品溶液,使铜质量浓度小于40mg·L-1后再进行上机测定。
2.6 试剂的干扰
试验引入的试剂,如硝酸影响较小,氢氟酸和高氯酸在测定前已被加热损失掉,少量硫酸可能影响待测元素的准确测定,因此试验考察了硫酸的干扰。在1.00,50.00μg·L-1混合标准溶液100mL中分别加入1~3滴25%硫酸溶液,按照仪器工作条件测定。结果显示,不加内标时3种待测元素的回收率为97.0%~106%,经内标校正后各待测元素的回收率接近100%,说明微量硫酸的存在不干扰待测元素的测定。
2.7 标准曲线和检出限
按照仪器工作条件测定混合标准溶液系列,以各待测元素的质量浓度为横坐标,对应的信号强度为纵坐标绘制标准曲线。结果显示,各待测元素标准曲线的线性范围均为0.20%~50.00μg·L-1,线性回归方程和相关系数见表7。
按照试验方法制备11份样品空白溶液,在选定的仪器工作条件下测定各元素含量,计算各待测元素测定值的标准偏差(s),以3s作为各元素的检出限,结果见表7。
表7 线性参数和检出限
2.8 标准物质和样品分析
按照试验方法分析标准物质 GBW 07168和两个锌精矿样品(编号分别为2#和3#),各平行测定11次,计算测定值的 RSD,并对这两个样品进行加标回收试验,计算回收率;同时参考文献分析样品2#和3#。所得结果见表8。
表8 精密度和准确度试验结果(n=11)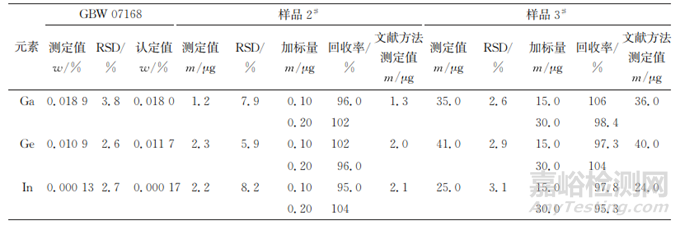
由表8可知:标准物质中3种待测元素的测定值和认定值基本一致,实际样品中3种待测元素的测定值和文献方法的测定值也基本一致,实际样品中3种待测元素的回收率为95.0%~106%,说明方法的准确度较好;测定值的RSD均 不大于8.5%,说明方法的精密度较好,满足锌精矿中镓、锗、铟的测定要求。
3、试验结论
本文采用硝酸-氢氟酸-高氯酸溶样体系分解样品,并在待测样品溶液中加入2滴25%硫酸溶液避免铁氧化物的析出;采用ICP-MS测定样品溶液中镓、锗、铟的含量,同时探讨了待测/内标元素同位素的选择以及质谱、非质谱和试剂干扰及消除。该方法简便、快捷,能用于锌精矿中镓、锗、铟的测定。
作者:谭秀丽1,左鸿毅2*,王文杰1
单位:1.深圳市中金岭南有色金属股份有限公司韶关冶炼厂;
2.深圳市中金岭南有色金属股份有限公司科学技术开发院
来源:《理化检验-化学分册》2024年第2期

来源:理化检验化学分册
关键词:
电感耦合等离子体质谱法