
嘉峪检测网 2024-10-13 10:59
导读:本课题组聚焦于自体CAR-T 细胞治疗产品药学变更案例及其共性问题与挑战,进行了行业调查、法规与案例收集和研讨以及变更管理工具评估,并提出了多项建议,期望为企业和监管机构管理CAR-T 药学变更提供参考。
摘 要
近年来我国在嵌合抗原受体T 细胞(CAR-T cell)治疗领域的研究非常活跃。由于该领域技术迭代快,生产工艺复杂,药学研究和变更方面的经验相对有限,CAR-T 产品药学变更的评估、研究以及申报、审评对企业和监管机构提出了多方面的挑战。本课题组聚焦于自体CAR-T 细胞治疗产品药学变更案例及其共性问题与挑战,进行了行业调查、法规与案例收集和研讨以及变更管理工具评估,并提出了多项建议,期望为企业和监管机构管理CAR-T 药学变更提供参考。相关法规收集与分析结果显示,我国和美国在法规方面均走在前沿,总体原则均为基于科学和风险;在细节和实践上,监管机构均处于持续研究、探讨和完善的阶段。行业调研结果显示,企业药学变更主要目的之一是降低成本以提高产品可及性,主要挑战包括企业和监管机构对变更理解的差异、沟通机制以及审评审批时长。收集的药学变更案例包括多种变更情形,其中针对扩大生产产能,建议基于适当产品与工艺知识经验、风险管控和无菌验证,可使用同步验证,适当时使用模拟验证辅助。就上市后变更管理工具,建议试行ICH Q12 批准后变更管理方案(PACMP)帮助管理自体CAR-T 细胞治疗产品上市后药学变更。
In recent years, research in the field of chimeric antigen receptor T (CAR-T) cell therapy in China has been active.Due to the rapid iteration of technologies in this field, complex manufacturing processes, and relatively limited experience in pharmaceutical research and changes, the evaluation, research, submission, and review of chemistry, manufacturing, and controls (CMC) changes in CAR-T products present various challenges for both companies and regulatory agencies. The Cell and Gene Therapy Products Committee of the China Society for Drug Regulation organized a task force comprising several leading domestic cell therapy companies to focus on case studies and common challenges related to CMC changes in autologous CAR-T products. The group conducted industry surveys, collected regulatory data and case studies, and evaluated change management tools, providing recommendations for companies and regulatory agencies to manage CMC changes for CAR-T products. Regulatory analysis revealed that both China and the United States are at the forefront of policy development,with general principles based on science and risk. However, both regulatory agencies are still exploring and refining details and practices. Industry surveys indicated that one of the main purposes of CMC changes is to reduce costs and improve product accessibility, with major challenges including differences in understanding between companies and regulatory agencies,communication mechanisms, and approval timelines. The collected CMC change cases involves various scenarios, including capacity expansion. For such changes, it is recommended to use concurrent validation based on appropriate product and process knowledge, risk control, and sterile validation, with simulation validation as an auxiliary when necessary. For postmarket change management tools, the study suggests piloting ICH Q12 Post-Approval Change Management Protocol (PACMP) to help manage post-market CMC changes for autologous CAR-T cell therapy products.
关键词
自体;嵌合抗原受体T 细胞;细胞治疗产品;药学变更;生产产能,验证策略;批准后变更管理方案
autologous; chimeric antigen receptor T cell; cell therapy products; CMC changes; production capacity; validation strategies; post-approval change management protocol
自从首款嵌合抗原受体T 细胞(chimeric antigen receptor T cell,CAR-T cell)治疗产品被美国食品药品监督管理局(Food and Drug Administration,FDA) 批准上市以来,CAR-T在血液肿瘤领域的临床治疗中取得了可观的成果, 全球的CAR-T 细胞治疗临床研究也得到了迅猛发展,尤其是美国和我国,其登记注册的临床研究数量远超其他国家和地区。经笔者统计, 全球已有10 款CAR-T 细胞治疗产品被批准上市,我国有5 款CAR-T 产品获批上市。
CAR-T 细胞治疗产品生产工艺新颖且复杂,技术迭代较快。目前CAR-T 细胞治疗产品市场仍不够成熟,为了提高CAR-T细胞治疗产品的可及性,在保证产品质量的前提下,研发企业不断优化生产工艺、扩大生产产能以符合市场需求,并不断探索对产品质量的理解,包括分析方法的优化、质量标准的变更等。无论是临床试验期间,还是产品上市后阶段,CAR-T 细胞治疗产品均面临频繁的药学变更。然而,不论是从研发企业对药学变更的实践角度,还是从监管法规对变更的管理角度,目前经验均相对有限,这给变更研究、申报及审评均带来了多方面的挑战。
在监管科学方面,虽然细胞和基因治疗产品技术评价与监管体系研究是中国药品监管科学行动计划的重点项目之一[1-2],但产业快速发展不断对监管体系的建设和完善提出更高要求,临床转化研究的规范性和科学性有待提高[1-4],药学变更的迫切需求对技术评价及监管也提出了新的挑战[5]。本文对比了国家药品监督管理局药品审评中心(Center for Drug Evaluation,CDE)和FDA 发布的药学变更相关指导原则,就药学变更关注点对行业进行了调研,重点收集了CAR-T细胞治疗产品的多种药学变更情形和来自企业界的变更实践案例,并就代表性案例进行了深入分析、提出了建议,以期为企业和监管机构管理CAR-T 药学变更提供参考。
1、 我国和美国的CAR-T细胞治疗产品药学变更相关指导原则比较
目前,全球主要国家或监管机构已经颁布了药品开发相关的系列监管文件,包括法规、指南、药品生产质量管理规范(good manufacturing practice ,GMP)以及一些地方性规范、团体规范等。但这些监管文件大多针对化学药品和传统生物制品,专门针对细胞治疗产品药学变更的法规文件并不多见。
近年来,我国努力推动建立和完善细胞和基因治疗产品监管体系,药品监管部门发布的先进疗法产品相关指导原则,如《免疫细胞治疗产品药学研究与评价技术指导原则(试行)》《体外基因修饰系统药学研究与评价技术指导原则(试行)》《体内基因治疗产品药学研究与评价技术指导原则(试行)》《细胞治疗产品生产质量管理指南(试行)》等,对于指导相关产品的药学开发具有重要意义。同时,多名行业研究者开展了相关研究,全面梳理了我国细胞治疗产业及监管体系现状,分析国外细胞治疗产品监管体系,并探究对构建我国监管体系的启示[6]。
CDE 在2023 年6 月发布了《自体CAR-T 细胞治疗产品药学变更研究问题与解答(征求意见稿)》,广泛收集社会各界的意见和建议,此后于2023 年11 月发布《自体CAR-T 细胞治疗产品药学变更研究的问题与解答》正式文件[7]。美国FDA在2023 年7 月,发布了《细胞和基因治疗产品生产变更及可比性》(Manufacturing Changes and Comparability for Human Cellular and Gene Therapy Products) 征求意见稿[8]。对比分析后发现,两个文件的定位不同。FDA 的文件是一个较高层面的指导原则,包含了管理、沟通和技术等多方面内容;CDE 的文件专注于药学变更的常见问题与技术要求,将变更研究的一般原则、细胞治疗产品变更研究的特殊性和可比性研究方案通过问答的方式进行阐述指导,并对8 个具体变更情形给出了技术解答、建议和指导,对企业开展药学变更与研究更具有实际的指导意义。基于此,笔者总结了几点思考:在现有指导原则和技术文件的基础上,考虑在适当的时候,起草和颁布更高层面的细胞产品药学研发与变更总体指导原则;在可比性研究中,阐述和明确药学可比是否充分,以及是否需要非临床或临床数据的支持;考虑和明确ICH Q12 变更管理工具对细胞产品的使用[9]。
2、 CAR-T 细胞治疗产品药学变更行业调研
基于细胞治疗产品药学变更的问题和挑战,课题组进行了行业调研,内容主要包括:变更管理一般问题、CAR-T 细胞治疗产品生产相关变更、体外基因修饰系统相关变更、质量控制相关变更、变更可比性、稳定性研究等。通过问卷调查共获得CAR-T 细胞治疗及相关领域的22 家公司反馈。从受访企业分布状况来看,涵盖了CAR-T 细胞治疗产品研发生产、设备试剂耗材供应以及合同研究组织/ 合同开发和生产组织(Contract Research Organization/Contract Development and Manufacturing Organization,CRO/CDMO)等上下游和服务环节,提示调查结果可在一定程度上反映CAR-T 细胞治疗领域普遍关注的挑战和难点。
对问卷调查结果进行分析后发现,企业开展药学变更的主要目的之一是降低成本以提高产品可及性。由此可见,企业面临的大部分变更都有可能是计划内的变更。对这类变更可采取前瞻性的管理方式。大部分受访者认为细胞治疗产品的变更具有挑战性,主要的挑战是企业和监管机构对变更理解的差异,其他的挑战还包括变更对产品安全性和有效性的评估、变更级别判定、变更沟通机制、变更审评审批时长等。因此,制定合理的可降低成本的变更计划、加深业界各方对产品质量及工艺的理解和与监管机构的沟通交流等对细胞治疗产品药学变更的评估和高效开展具有重要意义。
对于ICH Q12 建议的变更管理工具,44% 的受访者认为ICHQ12 变更管理工具对CAR-T 细胞治疗产品具有现实的指导意义;34% 的受访者了解ICH Q12 变更管理工具, 但是不清楚是否对细胞治疗产品具有指导意义;19% 的受访者不了解ICH Q12的变更管理工具。课题组认为,采用ICH Q12 建议的变更管理工具来管理CAR-T 细胞治疗产品的变更有利于提高企业在变更管理方面的水平以及与监管机构沟通的效率。但是,大部分的受访者不了解ICH Q12 推荐的变更管理工具或不清楚该工具对细胞治疗产品变更的意义。因此加强ICH Q12 变更管理工具的宣传和交流,对推动ICH Q12 变更管理工具在我国细胞治疗产品领域的实施具有重要意义。
3、 自体CAR-T 细胞治疗产品生产产能扩增变更验证策略
课题组从国内多家头部细胞治疗公司收集了18 个药学变更案例, 包括产能变更、场地变更、工艺变更、物料变更、检测方法变更和质量标准变更等多个方面。通过对变更案例进行分析整理,并对多个代表性案例进行深入剖析,以期了解相关企业对CAR-T 细胞治疗产品特定药学变更问题的认知和实践经验。本文仅就生产产能扩增案例进行阐述,结合实际经验提出针对产能扩增验证策略的建议,供企业与监管机构讨论与参考。
自体CAR-T 细胞治疗产品要求生产制备的独立性,即每个生产批次仅可制备唯一患者的样本。因此,细胞治疗产品的产能扩增经常是增加镜像生产模块或在同一生产模块中增加生产频次,同时生产工艺、流程、控制、物料、分析方法以及质量标准等均不发生变化。针对产能扩增,目前国内外采用的验证策略可大致分为传统验证、同步验证以及模拟验证3 类,其优缺点分析、法规支持以及相应的国内外案例或经验如表1 所示。
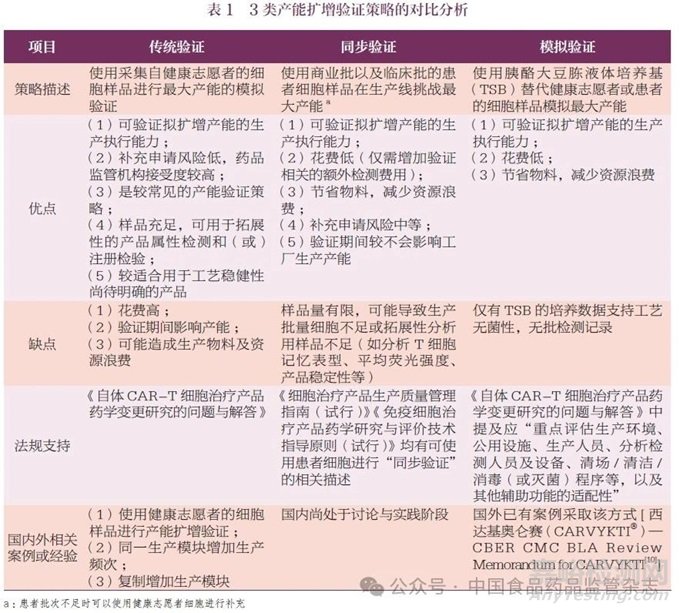
企业应根据产品所处的生命周期阶段以及产能扩增具体情况制定合理的产能验证方案。在确认工艺稳健性的前提下,结合已有产能验证经验,建议可采用同步验证的策略进行产能扩增,如有必要,可考虑结合模拟验证的方式。例如,针对已批准的生产模块,在生产工艺、物料、检测方法及质量标准均保持不变的情况下,仅通过增加生产频次进行产能扩增, 可考虑使用无菌工艺验证(Aseptic Processing Validation,APV) 挑战重点工艺步骤和操作时长,然后开展生产商业批和(或)临床批的同步验证。适当时,可通过风险评估来支持同步验证策略的实施,并通过后续GMP 所要求的APV 进一步支持。在开展产能扩增验证之前,建议企业与监管机构讨论确定验证策略、方案、标准以及在验证过程中生产样品的放行和用途。
对于在已批准的生产模块中仅通过增加生产频次的产能扩增,鉴于资源配置,通常采用逐级放大的研究策略。目前,针对此类变更仍需启动注册核查和注册检验,频繁的注册核查和注册检验对企业和监管机构都会造成较大资源占用,也可能影响患者可及性。建议监管机构针对该情形的首次变更进行现场核查,后续根据风险级别,决定是否启动注册检验和现场核查。
4、 使用PACMP 管理CAR-T 细胞治疗产品上市后变更的可行性分析
4.1 CAR-T 细胞治疗产品药学变更的监管挑战
针对自体CAR-T 细胞治疗产品临床研究期间的药学变更,目前已有完善的申报机制,一般由CDE 负责审评审批及监管[11]。对于上市后的药学变更,根据其级别和风险程度,由CDE 或省级药品监管部门受理或批准[12]。但目前省级药品监管部门对于新兴的细胞治疗产品尚难以进行备案,有些中等或微小变更也通常被要求向CDE 提出申请,从而将中等或微小变更作为重大变更进行对待,导致审评审批周期延长,部分甚至需要注册检验和注册核查,可能造成一些不必要的资源消耗,极大影响了变更的及时实施和产品的生产与供应。
4.2 ICH Q12 变更管理工具及在我国的实施情况
ICH《Q12 :药品生命周期管理的技术和监管考虑》关注产品生命周期管理, 引进了既定条件(Established Conditions,EC)、批准后变更管理方案(Post-Approval Change Management Protocol,PACMP) 和产品生命周期管理(Product Lifecycle Management,PLCM)等变更管理工具,以更具预测性和更有效的方式加强对上市后药学变更的管理[9]。在ICH Q12 实施之前,美国FDA 与欧洲药品管理局(European Medicines Agency,EMA) 就有类似PACMP 的概念和实践, 前者为可比性方案(Comparability Protocol,CP), 后者为PACMP。因此,美国FDA 在实施ICH Q12 初期试行的是EC 和PLCM, 而欧盟实施ICH Q12 时试行的是PACMP。
我国在该指导原则的准备与实施方面做了大量的工作。在过去5 年中,监管机构和企业举行了大量关于ICH Q12 的培训、研讨与行业会议,进行了广泛深入的国内外讨论与交流。2023 年4月26 日,CDE 发布了《关于公开征求ICH〈Q12 :药品生命周期管理的技术和监管考虑〉实施建议意见的通知》,就ICH Q12实施建议公开征求意见[13]。2023年8 月25 日国家药监局发布了《关于适用〈Q12 :药品生命周期管理的技术和监管考虑〉国际人用药品注册技术协调会指导原则的公告》[14]。公告指出,“申请人可以按照目前我国变更管理的相关法规规章和指导原则进行变更管理,也可以在提交上市申请和/或补充申请时采用Q12 提供的新方法进行变更管理。申请人在实施ICH Q12 前,应充分评估是否具备适用该指导原则的研发基础和实施条件。”同时鼓励在提交药品注册申请前与CDE 进行沟通交流。但目前尚无国内企业采用PACMP 管理CAR-T 细胞治疗产品上市后变更的先例。
4.3 使用PACMP 管理的可行性分析
在CAR-T 细胞治疗产品的开发过程中,企业对其工艺和产品的理解远不如传统小分子和大分子药物, 给EC 和PLCM 带来了困难。同时,基于CAR-T细胞治疗产品药学变更的挑战以及我国申报机制的特点, 建议先尝试采用PACMP 帮助管理CAR-T 细胞治疗产品的上市后药学变更。
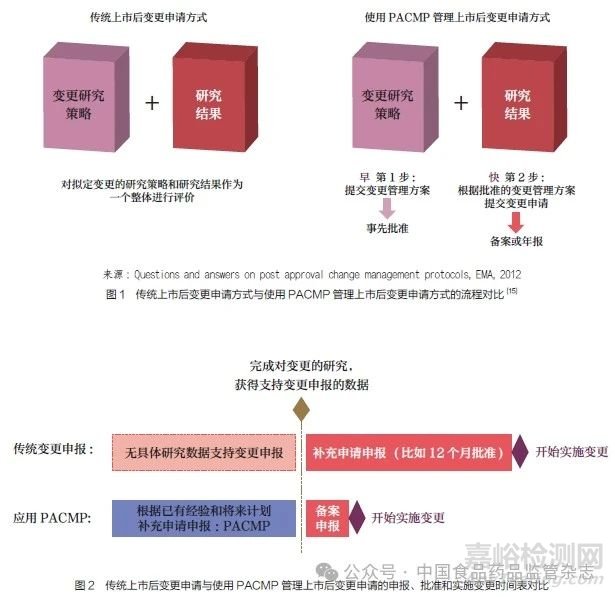
传统上市后变更申请方式与使用PACMP 管理上市后变更申请方式的流程对比如图 1 所示[15]。二者申报方式和研究时间线的区别导致了变更申报、批准以及实施的差异, 如图 2 所示。与传统上市后变更申请方式相比,PACMP 管理上市后变更的方式有助于加快变更实施进程,提高变更管理水平和效率,确保稳定的产品供应。
可使用PACMP 的药学变更种类包括中等变更和重大变更,例如:在同一生产场地扩大生产产能;增加病毒生产车间,使用相同的生产工艺和功能等同的生产设备;物料国产化替代变更;病毒载体有效期延长;细胞产品有效期延长;检测场地变更;制备病毒用细胞库变更;采血程序优化等。
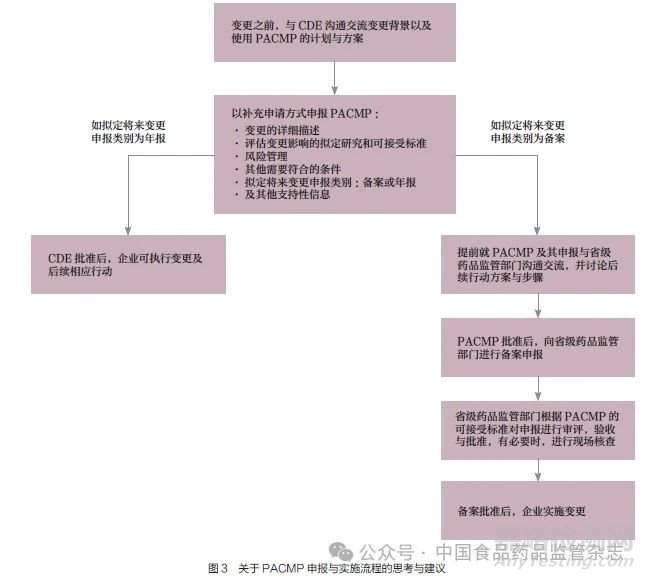
关于采用PACMP 管理CAR-T 产品上市后变更的申报与实施流程的思考与建议如图3所示。企业与CDE 沟通交流变更计划与方案并提出申请, 由CDE 对变更计划与方案进行审评审批。如拟定变更申报类别为年报,在变更计划和方案被批准后,企业可执行变更及后续相应行动。如拟定变更申报类别为备案,企业需提前与省级药品监管部门沟通交流,讨论后续行动方案与步骤并提出申请;待变更计划和方案被批准后,企业向省级药品监管部门进行备案申报;省级药品监管部门根据变更的可接受标准对申报进行审评、验收与批准。有必要时进行现场核查。待备案批准后,企业可实施变更。
如果采用PACMP 管理CAR-T 细胞治疗产品上市后变更,企业和监管机构除了关注申报与审评审批之外,还需要考虑注册检验与现场核查的问题。在注册检验方面,针对中等变更的情形,如果通过PACMP 以及后续的省级药品监管部门备案,是否可以避免不必要的注册检验;针对重大变更的情形,如果通过PACMP 申报批准, 以及后续必要的省级药品监管部门验收与核查,风险已经得到了管控,除常规注册检验之外,是否可考虑采用其他方式(如批次分析报告等)进行监测。在现场核查方面,针对中等变更的情形,如果通过PACMP 以及后续的省级药品监管部门备案,建议此类变更无需现场核查。如有必要,可通过年度检查等形式进行核查。针对重大变更的情形,在CDE 批准PACMP 之后,如省级药品监管部门接到申报备案并进行了验收,可考虑将现场核查作为验收的一部分,确保风险得到足够管控。
当然, 除了上述考量因素, 还有其他关键因素对保障PACMP 有效使用极为重要,如企业对产品的理解、变更研究设计、对ICH Q12 的理解与实践;监管机构对ICH Q12 PACMP 的理解与实施;各级药品监管相关部门之间的协调统一;企业与各级药品监管相关部门的充分沟通等。利用PACMP 管理不仅能充分评估和管控变更可能带来的风险,还能更有效利用相关资源,加速变更的实施,从而降低产品供应风险,尽可能满足患者的医疗需求。建议企业可提前就PACMP 及其申报与监管机构沟通,选择合适针对CAR-T 细胞治疗产品上市后变更项目进行试行。
5、 结语
基于CAR-T 细胞治疗产品的特殊性,其药学变更对企业和监管机构提出了多方面的挑战。本文聚焦于自体CAR-T 细胞治疗产品临床研究期间和上市后药学变更案例以及共性问题与挑战,进行了行业调查、课题研讨和思考。在法规方面,建议我国颁布细胞治疗产品药学研发与变更的总体指导原则,进一步阐明可比性是否需要非临床和临床数据的支持、变更管理工具(如PACMP)的使用、企业与监管部门沟通交流以及报告途径等事项,以更好地指导企业开展药学变更与研究。针对药学变更实践,本研究收集了18 个来自企业的实际案例,其中针对扩大生产产能的验证策略,建议基于相关产品与工艺知识经验、风险管控和无菌验证等考虑,可使用同步验证,适当时使用模拟验证辅助。此外,就上市后变更管理,本研究探讨并分析了采用PACMP 管理CAR-T 细胞治疗产品上市后变更的可行性,建议试行PACMP帮助管理自体CAR-T 细胞治疗产品上市后药学变更,并在实践中不断完善。
引用本文
曹晓平*,王武成,方淑平,王金辉,王永增,林右晨,封华,李付英,常桂红,周新腾,王国旭,王越*.自体嵌合抗原受体T 细胞(CAR-T)治疗产品药学变更实践与思考[J].中国食品药品监管.2024.09(248):16-25.

来源:中国食品药品监管杂志
关键词: 细胞治疗