
嘉峪检测网 2024-10-17 15:26
导读:当地时间10月16日,欧委会发布最新指南
当地时间10月16日,欧委会发布最新指南<MDCG 2021-25 Rev. 1:MDR 要求适用于 “遗留器械 ”和 2021 年 5 月 26 日之前根据指令 MDD和AIMDD投放市场的器械>
继21年MDCG 2021-25发布后,该指南首次调整,需注意的是,Rev.1版本对整个文件进行了调整,以与 MDCG 指南的一般结构保持一致(例如删除前言、工作组的任务和流程),并考虑到 2023 年 3 月 15 日颁发的过渡期延期条例((EU) 2023/607 ),该法规修订了MDR和IVDR法规关于某些医疗器械和体外诊断医疗器械的过渡性规定。
实质性修改尤其体现在 sections 3.1, 3.2 和 4,即
- 澄清MDR Article 19不适用于遗留器械;
- 澄清了过渡性条款对根据MDD Article 12(2)起草的声明所涵盖的系统和程序包的适用性;
- 澄清关于根据MDR Article 10(9)实施质量管理体系的要求。
1. 介绍
过渡期延期条例((EU) 2023/607 )对MDR的过渡条款进行了修订。特别是,过渡期已从 2024 年 5 月 26 日延长至 2027 年 12 月 31 日或 2028 年 12 月 31 日,具体取决于器械的风险等级并受某些条件的限制。
考虑到 MDR 过渡条款的修订,本文件就 MDR 要求对 “遗留器械”和 “旧/Old”器械”的适用性提供了最新指南。
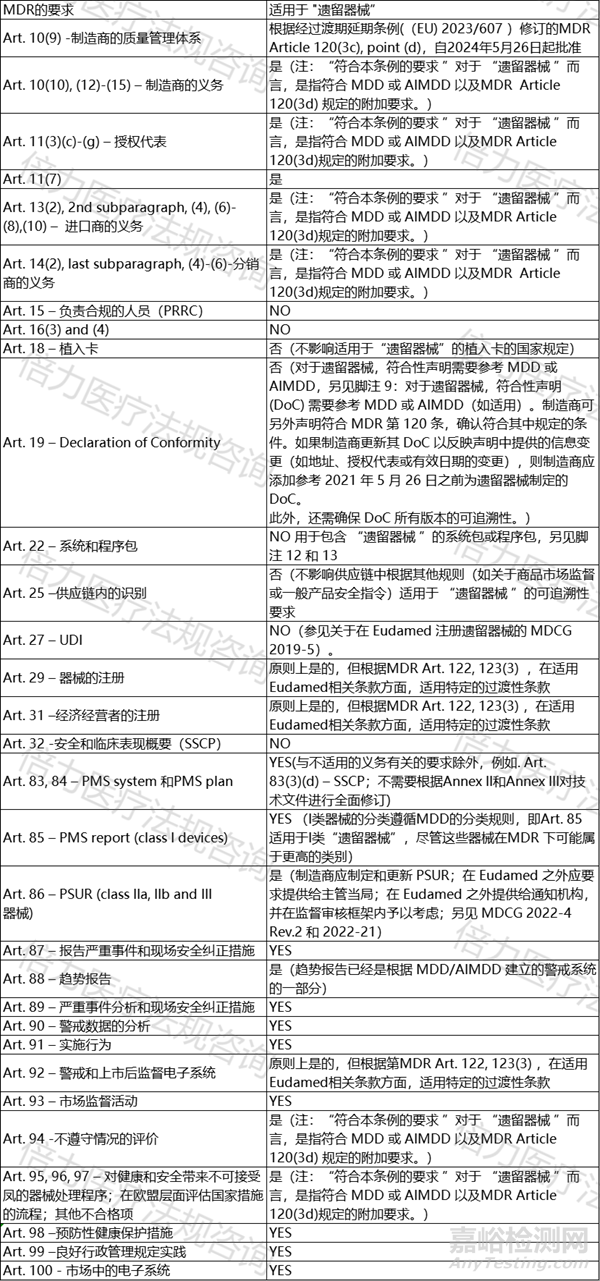
在文章最后的附录表格,说明适用于或不适用于“遗留器械”的MDR 要求。
2.法律规定和术语
2.1. 法律规定
经过渡期延期条例((EU) 2023/607 )修订的MDR Article 120(3)至(3e)条规定:

2.2. 本指南使用的术语
遗留器械应理解为根据MDR Article 120(3)的规定,在MDR的适用日期(DoA)之后、直至 2027 年 12 月 31 日或 2028 年 12 月 31 日(如果符合MDR Article 120(3c) 规定的条件)投放市场或投入使用的器械。这些器械可以是:
➢ 属于MDD指令规定的 I 类设备,在 2021 年 5 月 26 日之前已起草EU 符合性声明/DoC,且 MDR 规定的符合性评估程序要求NB参与的器械;
➢ 2021 年 5 月 26 日之前根据指令AIMDD或 MDD 颁发的有效 EC 证书所涵盖的器械。
旧/Old器械是指那些在 2021 年 5 月 26 日之前根据 AIMDD 或 MDD 或根据指令生效前的适用规则投放市场或投入使用的器械。
除 “遗留器械 ”外,MDR 器械是指符合MDR的投放市场或投入使用的器械。
需要提醒的是,投放市场的概念指的是每种产品,而不是某类产品。
3. 对 “遗留器械 ”适用 MDR 要求
3.1. 与上市后监督、市场监督、警惕性、经济经营者和器械注册有关的要求
根据MDR Article 120(3d) ,MDR中有关上市后监督、市场监督、警惕、经济经营者注册和器械注册的要求适用于 “遗留器械”。该规定与MDR最初版本相同,且未根据过渡期延期条例((EU) 2023/607 )进行修改。
这意味着MDR Chapter VII 中关于上市后监督、市场监督和警惕的所有相关MDR要求均适用于 “遗留器械”。
在确定取决于器械风险类别的要求(如MDR Article 85 or Article 86)的适用性时,应考虑根据MDR对原有器械进行的风险分类。根据MDR,其风险等级的可能变化仅与确定过渡期的结束有关。就过渡期内适用相关MDR要求而言,受MDR管辖的有源植入器械及其附件应被视为 III 类器械。
除了MDR Chapter VII 规定的要求外,MDR中与上市后监督、市场监督、警惕、 经济经营者和器械注册有关的其他要求也应适用于 “遗留器械”。
这种做法尊重了MDR Article 120(3d)的措辞。同时,它还将MDR的适用范围扩大到那些支持警惕性和市场监督系统良好运行以及经济经营者和器械适当注册的要求。
首先,制造商和进口商的一般义务是只将符合MDR的器械投放市场(MDR Articles 10(1) 和 13(1) ),而对于“遗留器械”,符合MDR 意味着符合MDD或AIMDD以及根据MDR Article120 (3d)的额外要求。此外,以下条款中规定的经济经营者的义务也应适用于经济经营者关于“遗留器械”的义务:
➢ 适用于制造商:Article 10(10), (12)-(15);
➢ 授权代表:Article 11(3)(c)-(g);
➢ 进口商 :Article 13(2), 2nd subparagraph, (4), (6)-(8), (10);
➢ 对于分销商:Article 14(2), last subparagraph,(4)-(6).
与上市后监督、市场监督、警惕、经济经营者和器械注册无关的MDR 要求原则上不适用于“遗留器械”的经济经营者/economic operators。
不适用 “遗留器械 ”的条款包括 Article 15, Article 16(3) and (4), Article 18, Article 19, Article 25, Article 27, Article 32。这并不妨碍经济运营商也对 “遗留器械 ”遵守任何 MDR 要求,特别是如果他们同时处理 “遗留器械 ”和 MDR 器械,并希望对所有器械采用相同的程序。
系统和程序包
按照MDR Article 120(2) and (3) 规定的过渡性条款的逻辑,过渡期也适用于系统和程序包:
- 根据MDR需要NB参与的系统和程序包
- 仅由 “遗留器械 ”组成,且
- 在 2021 年 5 月 26 日之前已根据 MDD Article 12(2)起草声明的系统和程序包
在这种情况下,MDR Article 22并不适用。根据系统或程序包中包含的MDR ,当最高风险等级的遗留器械的过渡期结束时,“遗留系统或程序包”的过渡期结束。这也适用于 2021 年 5 月 26 日之前由公告机构根据 MDD Article 12(3) 颁发证书的情况,除非该证书已被撤销。
根据MDD Article 12(2) 最后一项的规定,系统或程序包本身应被视为器械,但必须符合MDR Article 120(3)to (3d)规定的所有适用要求和条件,才能享受延长的过渡期。
3.2. 其他 MDR 要求
建立符合 MDR 的质量管理体系
根据 MDR Article 120(3c), point (d),制造商必须在 2024 年 5 月 26 日之前建立符合 MDR Article 10(9)的质量管理体系,才能获准在 2024 年 5 月 26 日之后将其 “遗留器械 ”投放市场。这意味着,自 2024 年 5 月 26 日起,制造商必须遵守MDR Article 10(9) 的规定,此外还必须遵守与市场后监督、市场监督、警惕、经济经营者和器械注册有关的规定,这些规定自 2021 年 5 月 26 日起已经适用。
对于遗留器械,QMS与MDR 的合规性不需要在2024年5月26日之前由NB认证,因为QMS的评估将由NB作为MDR 认证的一部分完成。
对于 MDR Article 10(9) 中列出的某些特定质量管理体系方面,例如 (b)、(e) 和 (f)点,需要考虑到质量管理体系涵盖 “遗留器械”、 即尚未(完全)符合 MDR 的器械。这意味着,对于这些器械,不要求制造商已确定所有相关的GSPR要求以及满足这些要求的方案,或已建立Annex I MDR Section 3 规定的风险管理系统,也不要求制造商根据 MDR Article 61 和Annex XIV 进行临床评估。
不过,制造商的质量管理体系应说明如何在过渡期内达到这些要求。
UDI 分配
由于 MDR Article 10(9), point (h)本身并没有规定 UDI 分配的要求,因此 UDI 分配的核查仅适用于实际需要对相关设备进行 UDI 分配的情况。如 MDCG 2019-5 所述,“遗留器械 ”不受 MDR UDI 要求的约束。这一方法在过渡期延期条例((EU) 2023/607 )中没有改变。
4.210526前投放市场旧器械适用MDR要求
涉及“旧/Old ”器械的严重事故和与 “旧/Old ”器械有关的现场安全纠正行动 (FSCA) 必须根据MDR Article 87 进行报告。
此外,MDR Articles 93 to 100 规定了主管当局在市场监督活动方面的权利和义务,也适用于 “旧 ”器械,因为这允许主管当局检查这些器械是否符合投放市场时适用的规则,并对不合规或不安全的器械采取适当措施。
适用于或不适用 “遗留器械 ” MDR 要求表

来源:Internet
关键词: 器械过渡期