|
NDA 代号
|
217639
|
|
申请人
|
Stemline Therapeutics, Inc.
|
|
剂型
|
片剂
|
|
规格
|
345mg和86mg
|
|
途径
|
口服
|
|
每日最大剂量
|
345mg
|
|
原研/OTC
|
原研
|
|
拟定适应症
|
ORSERDU是一种选择性雌激素受体拮抗剂,用于治疗内分泌治疗后疾病进展绝经后女性和男性,阳性、HER2阴性晚期或转移性乳腺癌,
|
|
产品描述
|
345mg:速释,浅蓝色,无刻㡾,椭圆形薄膜包应双凸片,一面印有“MH”,另一面无印字。
86mg:速释,浅蓝色,无刻㡾,椭圆形薄膜包应双凸片,一面印有“MH”,另一面无印字。
|
|
联合包装产品信息
|
无
|
|
包装信息
|
Elacestrant,红外薄膜包衣片剂、86mg和345mg包装在高密度聚乙烯(HDPE)瓶中,带有儿童防护(CRC)封口,并带有感应热密封衬。
|
|
储存条件
|
在20°C至25°C(68°F至77°F)下储存。允许在15°C至30°C(59°F至86°F)的偏移。[见USP控制室温]
|
企业Stemline Therapeutics寻求拟定的ORSERDUT (elacestrant)片剂批准,用于治疗-阳性、HER2阴性晚期或转移性乳腺癌。Elacestrant是一种选择性雌激素受体拮抗剂,在IND-124748下开发的新分子实体。Elacestrant 二盐酸盐是一种BCS 4类药物。拟定的Elacestrant片是一种速释制剂,未刻㡾,浅蓝色薄膜包衣片剂,含有86mg或345mg的elacestrant(游离碱)。支持本NDA的临床包括关键阶段3研究(RAD1901-308)和2个支持阶段1研究(研究005和研究106)和关键BE研究(RAD1901-116)。
本文主要内容来自于FDA的审评报告:PRODUCT QUALITY REVIEW(S),共32页。
基于PSD数据(参考模块3.2.P.2中表4)对于已用于临床批次片的API批次,PSD值的范围分别为D90 XX u m,D50 XX u m,D10 XX u m。拟定的溶出方法尚未展示出区分最小粒度原料药生产的片剂(D10:XX um,D50:XXum,和D90:XXum)和最大粒度原料药生的片剂(D10:XX um,D50:XXum,和D90:XXum)。尽得早期批次存在变异,但在粒度范围内原料药生产的所有临床批次片剂的溶出在30分钟都大于拟定的Q值。因此FDA建议原粒度范围设定在多批次均值上下3倍SD范围,如此体现临床批次用到的原料药粒度范围特性。
溶出方法验证
拟定的QC溶出方法已就色谱法进行了验证。溶出方法参数的耐用性(0.01 N HCI与XX N HCI)和桨速(XX rpmvs 75 rpm)在两种规格片剂得到了验证。在临床和注册/稳定性批次上,拟定溶出方法的溶出数据证明了方法的可重复性、可重现性和耐用性。
溶出标准
企业提定的溶出接受标准为Q=XX%(XX分钟)被认为是宽松的,不被FDA接受。在补充的资料中(SDN-29),企业澄清到,关键BE批次是2020年12月生产的,临床试验使用时间为2021年4月至7月。
在补充资料中(SDN-43)中,企业接受了FDA建议,并将溶出接受标准修订为“30分钟,NLTXX(Q)”。企业更新了P.5.1章节,包括新拟定的质量标准,FDA认为补充的资料是是充分的。
溶出方法和接受标准的临床相关性(例如,IVIVR,IVIVC,计算机模型,体内小范围)
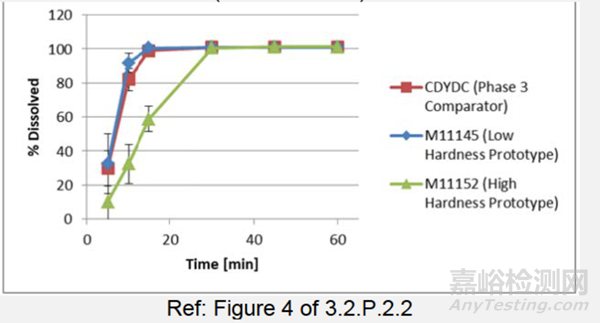
数据评估不同处方的PK:在PK研究(RAD1901-112)中,对两种具有不同片剂硬度的400mg拟商业配方进行了评估(表3)(在空腹或进食条件下),并与临床3期临床批次(在进食条件下)进行了比较。两种变更片剂的PK结果(AUC、Cmax、Tmax和T1/2)在空腹或进食条件下是一致的,与临床3期临床批次(在进食条件下)对比,其是满足生物等效性标准。低硬度拟商业片剂和临床3期片剂的溶出曲线图(图3)在15分钟内完全溶出,而高硬度拟商业片剂在30分钟内完全溶出。这表明30分钟前溶出差异不太可能影响体内性能,因为尽管片剂硬度不同,处方也发生了变化,但3种不同工艺批次被证明会产生类似的PK结果。
图3:Elacestrant 剂片,400mg,中试PK的低硬度和高硬度原型(处方E)及临床3期对比批次(配方D)的溶出曲线
表3:中试PK(RAD1901-112)原型(处方E,批次M11145和M11152)和临床3期对比批次(配方D:批次CDYDC)片剂的硬度,400mg
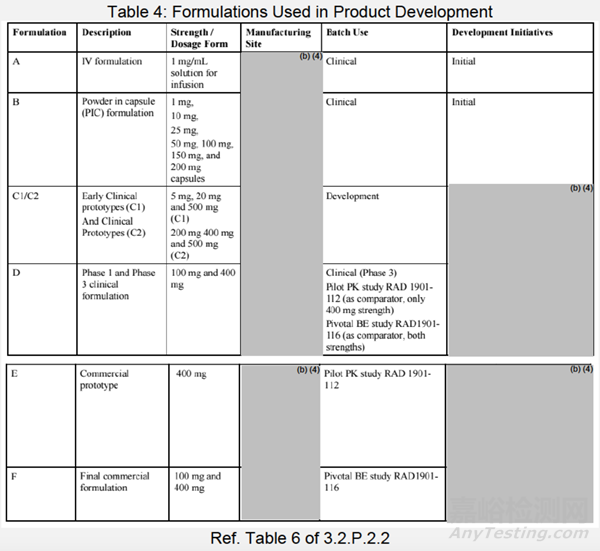
处方桥接
表4列出了所有临床试验处方的列表。该表概述了该药物开发过程中所做的变更,以及早期临床、1期和3期(处方D)、中试PK(处方E)和拟业商业化处方(处方F)之间的比较。最终的商业化处方(处方F)有不同的处方和生产场地。基于关键临床BE研究[RAD1901-116],建立了临床和拟商业化药物之间的桥接。
「解读」
尽管Elacestrant属于难溶性化合物,在可预见的溶出方法开发中,所有临床批次,其原料药粒度各有不同,但没有显示出可见的溶出区分行为,在30分钟都满足拟定的Q值。因此FDA建议,原料药的粒度接受标准使用多批临床批次的平均值加减3倍的SD值。
企业接受FDA的建议,修订了溶出方法Q值时间点,并更新了M3模块中相应的质量标准和方法章节内容。
企业同时对比了不同临床阶段,不同处方,以及不同硬度的片剂溶出曲线数据,发现低硬度,中等硬度的溶出曲线基本相拟,高硬度的溶出在前30分钟表现出明显的差异,但在30分钟都满足拟定的接受标准;上述三种批次在PK研究中没有表现出明显的差异,即PK数据相似。从中可以看出即使体外溶出表现出了不同的批次,体内PK数据没有差异,说明溶出表现与临床没有相关性,即溶出方法开发考虑为质量控制,即可满足方法要求,不必要考虑体内的相关性。

来源:Internet
关键词:
溶出