
嘉峪检测网 2024-11-12 11:01
导读:本文介绍了生物学评价相关的法规标准及实践中需要着重关注的点。
生物学评价一直是审评重点关注的问题之一,在CMDE中检索“生物学评价”你会发现有大量相关的法规及答疑,这是我们了解审评监管尺度的主要途径之一。今天,将和大家一起学习生物学评价相关的法规标准及实践中需要着重关注的点。
相关标准中的概念
生物相容性最主要也是最重要的标准就是GB/T 16866.1-2022/ISO 10993-1:2018,其中广泛的和相关标准中关于生物相容性的定义是这样的:
生物相容性:
生命体组织对非活性材料产生的一种性能。一般是指材料与宿主之间的相容性,包含组织相容性和血液相容性。
组织相容性:
细胞黏附性;无抑制细胞生长性;细胞激活性;抗细胞原生质变化性;抗炎症性;无抗原性;无诱变性;无致癌性;无致畸性。
血液相容性:
抗血小板血栓形成;抗凝血性;抗溶血性;抗白细胞减少性;抗补体系统抗进性;抗血浆蛋白吸附性;抗细胞因子吸附性
医疗器械生物相容性:
某一医疗器械或材料在特定应用中具有适宜宿主反应的能力(GB/T 16866.1-2022/ISO 10993-1:2018)
能够与一个生命系统接触而不会产生严重不良反应的能力(ISO 18562-1:2017)
2022版标准中新增了21个术语,给出了生物相容性的定义。历次版本的GB/T 16886.1中一直未给出生物相容性的定义,主要是因为生物相容性本身就是一个动态的定义,随着当前的认知水平不断更新。GB/T 16886.1-2022中正式给出了生物相容性定义,这一定义主要强调了两方面的内容:一方面是生物相容性离不开医疗器械或材料的特定使用条件。一种使用条件下具有良好生物相容性的医疗器械或材料在另一种使用条件下的结果可能大相径庭,比如PTFE材料用作人工血管具有良好的血液相容性,但是如果用作其它组织植入物则容易引起组织反应;另一方面,就是适宜的宿主反应能力。某一医疗器械或材料的宿主反应不是控制的越低越好,不应离开该器械或材料的总体设计,应兼顾其使用性能和安全性。同时,过度的控制也会增加生产成本,从而间接剥夺部分患者的使用权利。此外,在新型生物材料中,如可吸收组织诱导性生物材料,适宜的宿主反应可以诱导植入物的降解和修复组织的重塑和再生,这也是生物相容性需要关注的重要因素之一。
相关法规指南
最早在2007-06-27CMDE发布关于生物学评价的审查指南——《医疗器械生物学评价和审查指南》国食药监械[2007]345号,初步制定了生物学评价的审查重点,补充了评价方式和审评可接受度。后来在2018-11-13公开征求《医疗器械生物学评价指导原则第1部分:总则(征求意见稿)》意见,明确了生物学评价的基本要求、风险管理、评价过程和评价重点等信息。以上都是与GB/T 16866系列标准相辅相成的法规指南。
生物学评价的风险及过程在指导原则和标准中都有明确说明,这里需要再强调的是生物学评价的终点。
生物学评价的基本要求
在对新器械进行评价时,如果器械不与组织直接或间接接触(在GB/T 16866.1-2022中有相关的定义),应明确声明,且无需提供进一步的生物相容性信息。
对与人体直接或间接接触的医疗器械进行生物学评价,应遵循以下基本要求:
1、器械制造材料的选择及其生物相容性评价应首先考虑直接或间接组织接触的可能性以及任何关于器械制造的有用信息(例如,每项组件材料的化学配方,包括粘合剂、已知和疑似杂质、以及与加工相关的成分)。对于由纳米材料组成、包含纳米材料或会生成纳米材料的医疗器械,由于纳米材料潜在的特有性质,可能需要对其生物学评价进行特殊考虑。
2、另外,直接或间接接触医疗器械的包装材料可能将化学物转移到医疗器械上,然后间接转移给患者或者临床医生。因此,医疗器械的生物学评价还应关注产品的内包装材料可能引起的风险。
3、应考虑制造材料、成品、可能沥滤的化学物质或降解产物与器械总体毒理学评价的相关性。如有证据显示,特定的物理特性对生物相容性有影响,还应关注产品的物理特性。应关注的物理特性包括但不限于孔隙率、粒径、形状和表面形态。
4、与生物相容性评价相关的终点应考虑医疗器械与人体接触的性质、程度、频次、时间和条件。可利用该原则对器械进行分类,以便于在总体生物相容性评价中选择合理的终点。
5、境内申请人提供的医疗器械生物学评价报告中含有的生物学试验报告应由具有相应生物学试验资质的检测机构出具;境外申请人提供的医疗器械生物学评价报告中含有生物学试验报告的,应提供生物学试验室所在国的GLP证明。
6、应提供完整的试验数据,且此数据可用于独立做出结论。
7、应在医疗器械的整个生命周期内评估其生物安全性。对于可重复使用的产品,应对其最大验证处理周期数的生物安全性进行评估。
8、如果器械的化学成分、制造工艺、物理结构(如大小、几何构型、表面特性)、预期用途或初级包装发生任何变更,应针对生物相容性是否发生变化以及是否需要进行额外生物相容性试验的情况进行评价。
9、在评价器械改良情况时,如果改良不会变更任何与组织直接或间接接触的组件,申请人应明确声明,且无需提供进一步的生物相容性信息。但是,如果变更会对其他未经变更的、与组织直接或间接接触的器械部件造成影响,应进行生物相容性评价以确定变更可能造成的影响。例如,如果新增不与组织接触的内部组件,但是,需要通过加热使其与其它患者接触的组件相连,那么在加热时,可能影响与患者接触的组件,从而生物相容性也会受到影响,应进行评价。
10、在进行生物学评价时,应联合考虑其他非临床研究、临床研究和上市后经验信息,以整合所有有用的相关信息进行安全性评估。
生物学评价的终点概述
生物学评价的风险及过程在指导原则和标准中都有明确说明,这里需要再强调的是生物学评价的终点。详见下表
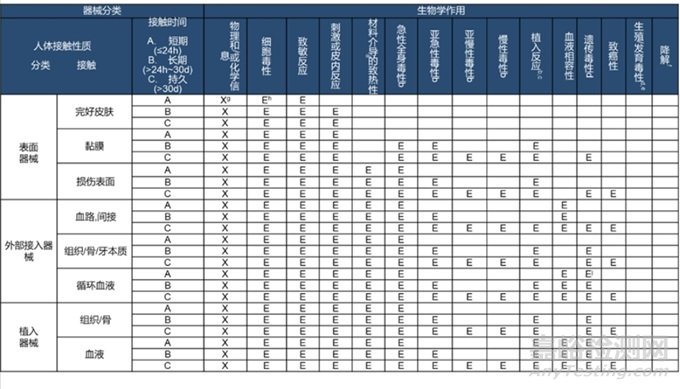
相关符号定义详见GB/T 16886.1-2022。
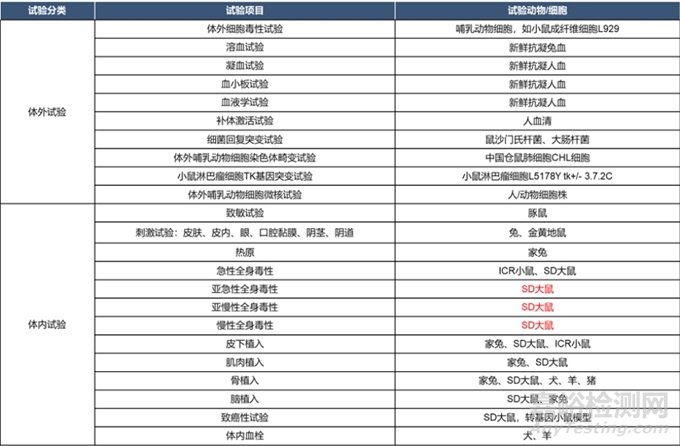
体内/外试验优先级
在进行体内动物试验之前优先进行体外实验,具体内容详见下表。

来源:医械铁锅炖
关键词: 医疗器械生物学评价