
嘉峪检测网 2024-11-13 20:54
导读:文章旨在从数量和关注的热点两个方面,对2023 年中国和美国发布的医疗器械指导原则进行比较分析,以揭示两国之间的差异,并据此为我国指导原则的体系完善提供相关建议。
内容提要:文章旨在从数量和关注的热点两个方面,对2023 年中国和美国发布的医疗器械指导原则进行比较分析,以揭示两国之间的差异,并据此为我国指导原则的体系完善提供相关建议。
关 键 词:医疗器械 指导原则 体系
美国医疗器械监管较早,经过长期的摸索已形成了较为成熟的监管模式,美国发布的医疗器械注册审查指导原则(以下简称指导原则)最早可追溯至1975 年[1]。美国食品药品监督管理局(Food and Drug Administration,FDA)多年在药品监管的科学性和创新性上保持领先,其经验和做法对于中国的药品监管体系具有重要的借鉴意义和学习价值。
1.数量分析
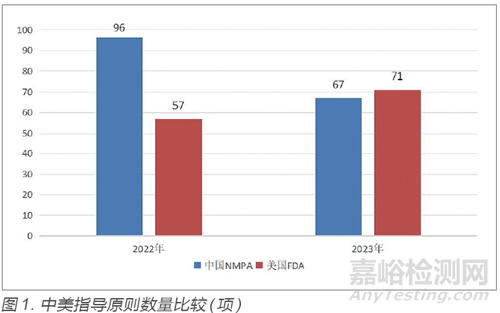
2023 年中国国家药品监督管理局(National Medical Products Administration,NMPA)共制修订67 项指导原则,相比2022 年较少了29 项(图1)。尽管指导原则数量有所减少,但2023 年中国NMPA共制定了222 项审评要点,围绕业内人士关注的医疗器械审评热点问题、开展满意度调查或座谈过程中收集到的需明确审评尺度的问题、与申请人/ 注册人沟通交流过程中收集的典型共性问题,发布了74 条共性问题解答。
2023 年美国FDA共制修订71 项指导原则,相比2022 年增加了14 项(图1)。指导原则数量的增加一方面表明美国FDA更加重视指导原则对医疗器械注册申报的指导作用,以帮助其更好地理解相关监管要求和流程;另一方面,从指导原则内容来看,美国FDA加强了对特定领域和产品的监管力度。
2.关注的热点问题
指导原则可以反映出监管机构关注的重点,审视美国FDA发布的指南文件,可以得出其关注的重点领域。
2.1 真实世界研究
当前,全球医疗器械创新速度加快,真实世界研究真实世界研究作为评估医疗器械安全性和有效性的重要手段,得到全球药品监管机构的重视和药械企业的关注,美国、英国、欧盟及中国在内,诸多国家围绕加强真实世界研究制定相关政策和指南。
2023 年12 月18 日,美国FDA发布了《如何使用真实世界证据(REW)来支持医疗器械监管决策》的草案,该指南草案是FDA对2017 年发布的定稿指南的更新,旨在澄清如何评估真实世界数据的相关性和可靠性、研究设计要素等,同时也为考虑使用真实世界证据来支持监管提交的医疗器械赞助商提供了更多的建议[2]。
2019 年中国NMPA开始全面开展REW用于支持医疗器械监管的工作,设立博鳌乐城国际医疗旅游先行区临床真实世界数据应用的试点[3]。在采纳国际医疗器械监管者论坛医疗器械临床评价工作组、登记数据库工作组成果文件的使用内容和参考境外监管机构技术指南的基础上,2020 年11月发布《真实世界数据用于医疗器械临床评价技术指导原则(试行)》。自启动海南临床真实世界数据应用试点工作以来,中国积极探索真实世界数据用于产品注册的路径,共有2批14个品种的临床急需进口医疗器械被纳入真实世界数据应用试点品种,共有7 个品种(9 个产品)已获批上市,2023年新获批3 个试点品种,共有10 个产品纳入真研前置沟通。
2.2 电子提交系统
随着科技的飞速发展,数字化的应用正在改变着传统的管理模式。医疗器械监管领域也不例外,各国医疗器械监管机构正在不断推动医疗器械的数字化,经过设计、开发、优化、测试等一系列过程,引入电子提交系统,以提升审评效率,加快审评速度,提高审评质量。
美国FDA于2018 年9 月启动了510(k) 质量审查计划试点,旨在简化制造商使用电子提交软件提交某些510(k) 通知的方式。经过试点计划、指南草案、开始试用、更新模板和一年的过渡期。2023年10月2日发布《医疗器械510(k)电子提交模板》指南文件,该指南明确2023 年10 月1 日后,正式启用eSTAR(即electronic Submission Template And Resource)系统,除非获得豁免,否则所有510(k)申请文件均需以eSTAR格式递交[4]。
中国NMPA2019年6 月24 日,正式启用医疗器械注册电子申报信息化系统(eRPS),开始实施电子申报制度,成为全球首个实现全项目、全流程注册申报电子化的国家。同时,发布了《医疗器械注册申请电子提交技术指南(试行)》,指导注册申请人通过医疗器械注册电子申报信息化系统进行电子格式申报资料的准备、提交和电子申请事项的管理[5]。截止2023 年底,医疗器械注册电子申报信息化系统累计接收各类注册申请事项5 万余件。
2.3 网络安全
医疗器械网络安全是医疗器械安全性和有效性的重要组成部分,也是国家网络安全的组成部分之一。随着网络技术的发展,越来越多的医疗器械具备网络连接功能以进行电子数据交换或远程控制,在提高医疗服务质量与效率的同时也面临着网络攻击的威胁[6]。医疗器械网络安全具有影响因素多、涉及面广、扩散性强和突发性高等特点,风险相对较高,因此需要加强相应监管工作,以保证医疗器械安全性和有效性,保障人民群众用械安全。
2023 年9 月美国FDA发布了《医疗器械网络安全:质量体系注意事项和上市前提交内容》最终指南,这一更新反映了FDA对医疗器械网络安全问题的高度重视,以及随着技术进步和市场变化而不断调整监管策略的需求。该指南更新了2022 年4 月8 日发布的同名指南草案,并取代了该机构2014 年发布的最终指南《医疗器械网络安全管理的上市前提交内容》,对开展网络安全风险评估、互操作性考虑因素以及向FDA提交的上市前材料中应包含的文件提供了更详细的建议[7]。
中国NMPA2022年3 月9 日发布了《医疗器械网络安全注册审查指导原则(2022 年修订版)》[8],该指导原则秉承着结合审评实际、借鉴国际网络安全监管最新成果的原则,对网络安全的基本概念、基本原则、网络安全与软件在生存周期过程方面的关系、技术考量、网络安全研究资料以及注册申报资料说明进行了全面和细致的描述和要求。但相比美国FDA网络安全指南,从内容上,涵盖了美国FDA相关指南的核心内容。囿于行业发展水平,在网络安全监管方面中国NMPA与美国FDA还有一定差异。
2.4 突破性器械项目
为了提升医疗健康水平、推动产业升级、促进更多创新医疗器械上市,各国通过启动项目和计划、优化审批流程与机制、发布相关指南、提供政策支持和指导、加强国际合作等多维度、全方位的举措,增强自主创新能力。
突破性器械项目是美国FDA于2015 年启动的加快创新医疗器械开发和审查过程中的“绿色通道”,旨在更快地将那些为了更有效地治疗或诊断生命威胁或不可逆转地破坏人类健康的疾病或病症的设备和产品引入市场。2023年9月美国FDA更新了《突破性医疗器械程序》指南,以明确一些促进健康公平或治疗疼痛的非成瘾性的产品也可能适用于突破性程序,这一更新进一步拓宽了突破性器械项目的适用范围,鼓励更多具有创新性和临床价值的医疗器械进入市场。2023 年美国FDA共批准124 个突破性医疗器械上市[9]。
2018年中国NMPA废止了2016年发布的《创新医疗器械特别审批申报资料编写指南》,发布了新的《创新医疗器械特别审查申报资料编写指南》,以期提高申报资料质量,使申请人明确在申报过程中应予关注的重点内容,解决申报过程中遇到的一些共性问题。2023 年中国NMPA共批准了61个创新医疗器械上市,呈现出产品多样、技术突出、自主知识产权、临床应用价值高等特点,但在创新能力、技术水平、法规要求方面,中国与美国还存在一定的差距,这既是挑战也是未来发展的动力。
2.5 人因工程
人因工程是医疗器械可用性和安全性的系统工程,是医疗器械安全、有效性的基本要素之一,也是全球主要监管体系日趋重视的风险点之一。美国、英国、欧盟以及中国等监管机构都已经制定了相关要求,要求企业在其整个医疗器械产品开发过程中严格执行人因工程,最大限度地降低使用错误的可能性,以提高医疗器械的安全性和有效性。
美国FDA通过发布一系列指南和草案,不断规范和完善人因工程原则应用的相关要求,2022 年12 月公布《医疗器械人因申报指南》草案,2023 年9 月7 日发布《药品器械组合产品人因工程原则应用:问题与解答》草案,进一步明确了人因工程原则在组合产品研发各阶段的考量要点,包括产品设计、临床前研究以及上市前验证,以促进组合产品的开发[10]。
中国NMPA是2019 年将《医疗器械人因设计注册审查指导原则》列为指导原则制定计划,通过成立工作组、召开启动会、确定章节架构、形成初稿、征求意见、召开定稿会、参考FDA公布的人因申报指南征求意见稿、开展调研等一系列工作,2023 年10 月11 日第二次公开征求《医疗器械人因设计注册审查指导原则(征求意见稿)》。目前,《医疗器械人因设计注册审查指导原则(征求意见稿)》已上报至中国国家药品监督管理局医疗器械注册管理司。
3.相关思考及探讨
在真实世界研究、电子提交系统、网络安全、突破性器械项目以及人因工程方面,中美两国的指导原则既有差异性又有共同关注的核心议题。为了推动医疗器械产业的高质量发展,我国应持续完善我国指导原则体系,提升指导原则的科学性、前瞻性和引领性。
一是增加通用类指导原则,构建产品类和通用型(模块)互补的指导原则体系,为不同类型、不同阶段的医疗器械研发与生产提供更为精准和实用的指导。二是仍需密切关注美国、欧盟、日本等发达国家和世界卫生组织、国际医疗器械监管者论坛等国际组织的最新技术指南和最佳实践,结合我国审评审批工作实际及时进行转化应用,为医疗器械产业的高质量发展提供有力支持[11]。三是医疗器械监管正朝着数字化、高效化和创新化的方向发展,根据工作实际需求,基于业务领域关注的前沿技术或热点领域推荐,或通过人工智能医疗器械创新合作平台、生物材料创新合作平台,增加发布风险高、数量集中、行业关注度高的指导原则力度。通过及时发布具有前瞻性的指导原则,引导企业合理布局研发方向,规避潜在风险,促进技术创新与产业升级。同时,对于已发布的指导原则,应建立定期评估与修订机制,通过持续跟踪产业发展趋势和监管需求变化,及时对指导原则进行修订和完善,确保其始终与产业发展实际保持高度一致,避免指导原则与产业实际脱节。
面对日新月异的技术进步和新产品不断涌现的挑战,我国仍要继续秉持审评科学的核心理念,紧跟科技创新的发展趋势,密切关注国际形势中的风险考验,开发健全新的审评工具和新的审评方法,来科学评估产品的安全、有效、质量和性能,推动对科学审评的认识与应用,以产品为核心、以科学证据为基础、以指导原则及标准为工具开展审评工作,促进审评科学理念的统一与执行。

来源:中国医疗器械信息
关键词: 医疗器械指导原则