
嘉峪检测网 2025-02-08 08:31
导读:本文主要从以下2方面阐述如何开展起始物料的质量研究话题:申报资料的法规要求汇总;质量研究的一般要求.
原料药的起始物料(下文以SM表示)为原料药的重要结构片断被引入,同时也是GMP监管的起点。
起始物料的质量间接关乎到原料药的质量。近些年来,原料药注册对于起始物料的研究要求,越来越严格。
本文主要从以下2方面阐述如何开展起始物料的质量研究话题:
● 申报资料的法规要求汇总
● 质量研究的一般要求
1、申报资料的法规要求汇总
SM需要递交的研究内容,主要来源于ICH M4Q 3.2.S.2.3物料控制章节中如下要求:
3.2.S.2.3 物料控制(名称、厂商)
Materials used in the manufacture of the drug substance (e.g., raw materials, starting materials, solvents, reagents, catalysts) should be listed identifying where each material is used in the process. Information on the quality and control of these materials should be provided.
应列出API生产用物料(如,原材料、SM、溶剂、试剂和催化剂)并说明在工艺中哪一步使用了这些物料。应提供这些物料质量及控制的信息。
上述M4Q主要为框架式要求,更为详细的还需看如下一些法规和指南中3.2.S.2.3的要求:
a)CDE《化学药品新注册分类申报资料要求(试行)》2016年80号文
3.2.S.2.3 物料控制
按照工艺流程图中的工序,以表格的形式列明生产中用到的所有物料(如SM、反应试剂、溶剂、催化剂等)的相关信息,并说明所使用的步骤。示例如下:
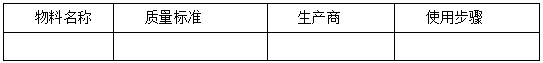
提供以上物料的质量控制信息,明确引用标准,或提供内控标准(包括项目、检测方法和限度),并提供必要的方法学验证资料。
对于外购的起始原料,为避免对API的质量引入不可控因素,需要提供起始原料生产商出具的制备工艺,并根据相关技术指导原则、技术要求对杂质进行全面的分析和控制,明确可能对后续反应影响的杂质或可能引入终产品的杂质(如,无机杂质、有机杂质、有机溶剂等),采用适当的(必要时经规范验证的)分析方法进行控制,根据各杂质对后续反应及终产品质量的影响制订合理的内控标准。
结合API的制备工艺要求、起始原料生产商提供的制备工艺和控制标准制定起始API的内控标准,说明内控标准(尤其是杂质限度与含量)的制定依据。
b)1EDQM: Content of the dossier for CEP applications for chemical purity and microbiological quality of substances for pharmaceutical use (2024)
Control of materials (3.2.S.2.3):
The impurity profile of starting material should be sufficiently understood and described. Any limitation in understanding the impurity profile of a starting material should be explained and justified along with a discussion on the impact on the impurity profile of the final substance. The specifications should reflect the synthetic strategy adopted and should include acceptance criteria for purity and/or assay, as well as impurities (specified, unspecified and total impurities, residual solvents, reagents including daughter-compounds, elemental impurities and mutagenic impurities), as needed.
Acceptance criteria should be justified by information on fate and purge of impurities, supported by data as needed.
应充分了解和描述SM的杂质谱。对于起始材料杂质谱的任何限度,都应进行解释和论证,并讨论对最终物质杂质谱的影响。质量标准应反映所采用的合成策略,并应包括纯度和/或含量的可接受标准,以及所需的杂质(特定的、非特定的和总杂质、残留溶剂、包括子成分在内的试剂、元素杂质和致突变杂质)。
可接受标准应基于杂质的去向和清除信息,并根据需要提供数据支持。
c)2EMA(Guideline on the chemistry of active substances:EMA/321776/2024)
Control of materials (3.2.S.2.3):
Active Substance (AS) Starting Material(s)
Complete specifications should be provided, including limits for impurities. The possibility that any kind of impurity, for example isomeric impurities or mutagenic impurities (including those from the ‘cohort of concern’, Ref 10), present in a starting material may be carried through the synthetic process unchanged or as derivatives should be discussed. Such impurities should, if relevant, be controlled in the starting material by appropriate acceptance criteria with suitably validated methods. Acceptance criteria should be established by the applicant based on evaluation of the fate of impurities present in the starting material, when subjected to the normal processing conditions.
Risk of formation and carry-over of nitrosamines during the starting materials synthesis should be evaluated (e.g., use of nitrosating agents, secondary or tertiary amines, etc.) (Ref 11). If a risk is identified, adequate control strategies (in the specification of the starting material or further downstream in the active substance process) should be established.
应提供完整的质量标准,包括杂质限度。SM中存在的任何种类的杂质,例如异构杂质或诱变杂质,可能在合成过程中保持不变或作为衍生物进行讨论。如果相关,应通过适当的可接受标准和适当验证方法控制SM中的此类杂质。
可接受标准应由申请人根据对SM中杂质在正常生产下的去向来确定。
应评估SM合成过程中亚硝胺形成和残留的风险。如果发现风险,应制定适当的控制策略(在SM质量标准中或活性物质工艺的下游)。
综上所述,总结下在申报资料里需要递交哪些内容涉及到质量研究和控制策略:
1.SM的质量标准
2.SM的质量标准的制定依据
3.SM的分析方法以及验证
2、质量研究的一般要求
SM涉及的质量研究内容,其实是一个原料药质量研究的简化版。下文将结合常见的一些发补要求,对于上述需要递交的内容,展开阐述:
1、SM的质量标准
API的质量标准和可接受的限度,可以参考各国的主流药典(EP,USP,JP,BP),同时按照ICH Q3A (针对特定和非特定), Q3C(残留溶剂),Q3D(元素杂质), M7(致突变杂质),EMA&FDA(相关亚硝胺指南)展开质量研究。
通常设置的项目有性状,鉴别,有关物质,异构体,致突变杂质,残留溶剂,炽灼残渣,水分/干燥失重,含量.
那么SM呢?
可参考API,酌情删减,设置为如下几个质量标准控制的项目
通常设置的项目有性状,无机杂质盐类,鉴别,有关物质,异构体,致突变杂质,水分/干燥失重,含量
2、SM质量标准的制定依据
这里为质量研究的重点内容,哪些项目经过质量研究后,需要纳入质量标准;哪些项目是仅做质量研究,不需要纳入质量标准。
主要从以下几个方面,阐述:
● 性状以及理化项目
● 有机杂质
● 致突变杂质
● 残留溶剂
● 无机杂质
● 元素杂质
2.1 性状以及理化项目
性状(如外观、颜色、物理状态)、理化项目(熔沸点、比旋度、pH等)。
一般可以参考供应商的标准以及多批次的实际情况制定内控标准。
2.2 有机杂质
2.2.1 SM中杂质的来源
可以同样参考API的杂质研究思路。
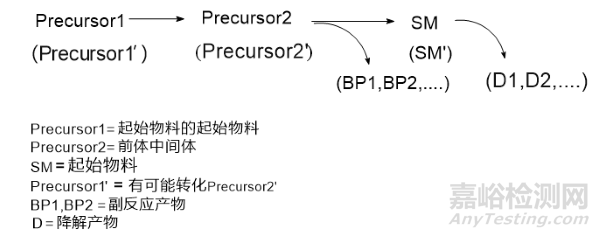
通常需要根据供应商提供该物料的简要的合成工艺(包含溶剂,试剂,催化剂),物料COA,再结合文献和反应机理,对可能存在的杂质进行了理论分析,建立SM的杂质谱。再根据检测,确认实际存在的杂质,排除不存在的杂质。
可能存在的有机杂质来源一般有如下图,可能包含未反应的物料,副产物,衍生杂质。
重点关注:同系物,异构体,异位取代,脱卤素杂质,同时关注警示结构杂质。哪怕审评机构会在发补阶段,还会要求补充一些杂质(对比同品种后)。
2.2.2 SM中杂质的限度
SM中的一般有机杂质主要分为:特定杂质,非特定杂质。
API中非特定限度一般参考ICH Q3A的鉴定限来制定,特定杂质按照界定限和毒理数据制定,如下图。
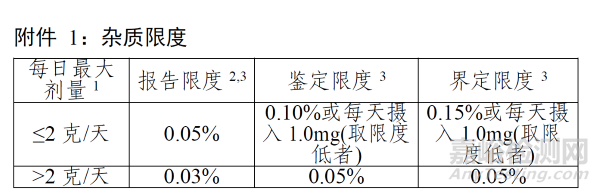
对于SM中杂质限度制定问题,通常有如下常见发补要求:
常见发补有:
(1)将含量较高的未知杂质作为特定杂质控制,收严其他单杂限度。
(2)根据多批次杂质实测值收严杂质的限度。
对于上述发补,主要:
● 因申请人参考供应商单杂限度,制定了SM内控的单杂水平。
而往往供应商的单杂限度较高(如1.0%或0.5%以上),需要提供含量较高杂质的去向。
可按照对应的API的界定限来制定非特定杂质限度。
● 因申请人参考供应商的特定杂质(或者单杂)限度,制定了SM内控的特定杂质(或者单杂)限度。
虽然在制定特定杂质限度时,可参考供应商标准中的特定杂质水平制定内控标准。
虽然可反映供应商工艺控制该杂质的一般水平,但供应商实际检测结果往往是远小于其制定的限度。
那么如果按照供应商的限度来制定SM的内控限度,即使最后生产出合格的API,也只能表明含杂质(实际水平)的SM,能生产出到合格的API。不能表明当SM含有供应商质量标准中水平(可接受标准的宽的水平),也还可以生产出合格的API。
举例:供应商单杂质标准为(1.0%或0.5%),实际多批次检测结果均为:约0.1%。
用该实际水平(约0.1%)的SM生产出合格的API,只能该水平满足需求。
不能说明含有(1.0%或0.5%)水平,也同样可以生产合格的API。
● 不想根据实测结果收紧限度怎么办?
如果根据实测结果收紧限度,那么势必会对后期增加SM供应商有一定限制。
为了后续新增供应商以及供应链考量,往往需要加标试验来证明较高限度的合理性。
供应商单杂的限度,是一个重要的参考点。假设供应商标准单杂为X,通常可以通过杂质加标1.2X-2.0X(主要基于放大生产的杂质清除效果差一些和检测误差)试验获得工艺清除因子,SM中的杂质在API的限度(若不需要报告),倒推SM中该杂质限度.
举例:供应商单杂限度为X(如0.5%)。SM中某一个特定杂质Y(实测: 0.3%),按2X加标. 后续中间体1中,杂质Y残留水平为0.06%,即清除因子1.0%/0.06%=16.7. 若满足后续不在控制,Y的限度在SM里可制定为0.05%*16.7*80%=0.66%(80%安全系数)
2.2.3 SM中杂质的去向
SM中杂质的质量研究的另外一个重点即去向。
结合最终API最大日剂量来按照ICH Q3A报告限。尽可能的早闭环,避免滚雪球,越滚越大。
通常要求SM中杂质不能影响杂质谱即不能高于鉴定限(持续存在的杂质除外),否则延伸到SM的另外申报要求即认为SM的选择不合适。
☆ SM中实际低于鉴定限(0.10%或0.05%)的杂质,无需过多关注,避免过度研究。
☆ SM中含量较大杂质诸如高于鉴定限(0.10%或0.05%)以上的杂质,需要了解其在后续传递携带情况。
在后续中间体/API中杂质水平低于报告限即为去向的终点。
当杂质在API中的残留水平低于报告限,即使API的分析方法无法检测到(检出),也不需关注,不需要新增检测方法(增加额外的检测)
2.3 致突变杂质
首先通过文献,M7附录,CPDB & Lhasa Carcinogenicity等毒理数据库,来查找TD50值.看是否有可用的数据。
● 若有,根据API的每日最大剂量来计算出该杂质在API的限度。
● 若无,可需要通过QSAR软件评估确定最终分类,需要根据给药时长和API的每日最大剂量按照TTC计算该杂质在API的限度。
因致突变的限度往往都比较低,对仪器和方法的灵敏度有一定的要求,一般很少在SM中按照低灵敏度的方法检测。
SM到最终的API,还有较长的步骤,通常后续工艺有一定的清除效果。往往会根据清除因子(或清除率)来制定SM中的致突变杂质的控制限度,即控制方法3 和4。
|
分类 |
拟定的控制策略 |
|
1&2&3 |
根据给药时长和API的每日最大剂量,按照对应的TD50或TTC 计算限度。
a) 在SM中按照该限度订入质量标准.
b) 按照多梯度的限度,进行加标做清除试验,在API中低于30%TTC的限度或,在SM中按照加标限度,订入质量标准.
c) 按照10%TTC的限度或者多梯度的限度,进行加标做清除试验.或按照理论清除因子,来确认API中该杂质残留水平.
若三批实测检测结果低于限度 10% 或理论清除1%TTC以下。均可不订入质量标准。 |
|
4&5 |
按照一般杂质控制 |
亚硝胺杂质的研究,在SM中一般同致突变杂质类似。
其控制策略也是类似,此处不做过多阐述..
一般杂质和致突变杂质的来源,去向,控制策略,如下图,可供参考:
|
方法 |
杂质名称 |
来源 |
多批次SM中杂质 |
控制策略 |
|
方法1
(有关物质) |
SM-Imp1 |
未反应的物料。
不参与后续反应 |
≤ IT
(0.05%或0.10%) |
按照非特定杂质控制,限度不得过0.10% |
|
SM-Imp2 |
未反应的物料。
不参与后续反应 |
≥ IT
(0.05%或0.10%) |
多批次SM中杂质SM-Imp2约为:0.1%~0.2%.
验证3批中间体1中SM-Imp2均未检出(<RT)。
在SM中设定限度为0.2%,按照特定杂质控制. |
|
|
SM-Imp3 |
未反应的物料。
不参与后续反应 |
≥ IT
(0.05%或0.10%) |
多批次SM中杂质SM-Imp3约为:0.1%~0.2%
验证3批中间体1中SM-Imp3未检出(LOD: <RT)。
进行0.5%的加杂试验, 中间体1中SM-Imp3未检出,
在SM中设定限度为0.5%,按照特定杂质控制. |
|
|
SM-Imp4 |
工艺杂质。
参与后续反应,
转化成INT1- Imp4. |
≥ IT
(0.05%或0.10%) |
多批次SM中杂质SM-Imp4约为:0.1%~0.2%.
验证3批中间体1中SM-Imp4和INT1-Imp4均未检出(LOD: <RT)。
进行了0.5%的加杂试验。中间体1中SM-Imp4和INT1-Imp4均未检出(LOD: <RT),
在SM中设定限度为0.5%,按照特定杂质控制. |
|
|
方法2
(致突变杂质) |
SM-Imp5 |
未反应的物料。
参与后续反应 |
≥ IT
(0.05%或0.10%) |
其具有警示结构,API中可接受水平为:150ppm
多批次SM中杂质SM-Imp4约为:0.1%~0.2%.
验证3批,中间体1中SM-Imp4和INT1-Imp4均未检出(LOD=1ppm,LOQ=3ppm)。
进行了0.5%的加标试验。中间体1中SM-Imp4和INT1-Imp4均未检出,按照ICH M7 option3,
在SM中设定限度为0.5%,后续不做进一步控制 |
|
方法3
(致突变杂质) |
SM-Imp6 |
未反应的物料。
不参与后续反应 |
≥ IT
(0.05%或0.10%) |
其具有警示结构,API中可接受水平为:150ppm
多批次SM中杂质SM-Imp4约为:0.1%~0.2%.
验证3批,API中SM-Imp4均未检出(LOD=1ppm,LOQ=3ppm)。
进行了0.5%的加标试验。中间体1中SM-Imp4和INT1-Imp4均未检出,按照ICH M7 option3,
在SM中设定限度为0.5%,按照该限度开发高限度的方法检测。
API不做进一步控制. |
|
方法4
(仅质量研究) |
SM-Imp7 |
机理推断的潜在的杂质 |
≤ IT
(0.05%或0.10%) |
不足进行一步控制,仅做质量研究考察. |
2.4 残留溶剂
从SM开始至最后的API,可能会使用其他的溶剂,后续的工艺各种后处理步骤,且API会结合溶剂使用(引入)的步骤和残留水平来进行残留溶剂的控制,一般SM中不予重点关注,即使SM有1类溶剂的使用。
2.5 无机杂质
根据SM合成工艺,如果使用了无机盐类,采用炽灼残渣进行控制;还需要结合API工艺的清除情况,诸如后续会有大量的水洗等操作,无机杂质的限度可以制定更高。
2.6 元素杂质
SM到API可能还会引入其他元素杂质,那么对于元素杂质的研究,通常会在最终的API研究。
对于SM中使用特殊元素,一般无需在SM中进行研究,往往会在最终API中根据ICH Q3D进行该元素杂质研究。
3、起始物料的分析方法以及验证
对于SM中的分析方法,需要结合杂质的限度,开发适用的方法。
对于方法的验证,通常会对于需要追踪去向的杂质,因需要知道杂质去向。
需要定量验证,而对于表观水平较低(仅推测,实际未检出)杂质,可限度验证即可。
参考文献:
1.Content of the dossier for CEP applications for chemical purity and microbiological quality of substances for pharmaceutical use:2024),
2.Guideline on the chemistry of active substances:EMA/321776/2024)

来源:注册圈
关键词: 原料药起始物料