
嘉峪检测网 2025-05-10 10:34
导读:本文介绍了Crinecerfont胶囊CMC部分评估。
|
药品名 |
CRENESSITY (Crinecerfont) |
|
剂型 |
胶囊 |
|
规格 |
25 mg, 50 mg, 100 mg |
|
使用途径 |
口服 |
|
Rx/OTC |
Rx处方药 |
|
适应症 |
适应于糖皮质激素替代的辅助治疗,以控制4岁及以上患有经典先天性肾上腺增生的成人和儿科患者的雄激素 |
|
最大日服用剂量 |
200mg |
Crinecerfont属于新化学分子,其特性鉴定、纯度、杂质及生产工艺均通过适当质量管理体系和验证的分析方法得到有效控制。原料药在商业包装内于规定贮存期内稳定性良好。
该新药为口服软胶囊剂型,辅料成分包括:中链甘油三酯、二辛酸/二癸酸丙二醇酯、月桂酰聚氧-32甘油酯及维生素E聚乙二醇琥珀酸酯。需说明的是二辛酸丙二醇酯虽首次应用于口服制剂,但根据药学/毒理学审评意见确认其在处方中的定量水平可接受。药品规格分为25mg、50mg和100mg三种,采用儿童防护盖的HDPE瓶包装。生产过程由cGMP合规生产设施的质量管理体系有效管控。从质量角度评估,拟定的控制策略能够充分保证产品在特性、规格、纯度、效价及稳定性方面的质量一致性。
原料药部分评估
Crinecerfont分子中含有一个S构型的手性中心。表征数据确保了原料药的同一性和纯度。原料药结构已通过核磁共振氢谱(1H NMR)、核磁共振碳谱(13C NMR)、元素分析、傅里叶变换红外光谱(FTIR)、质谱(MS)以及单晶X射线衍射分析得到充分表征。立体中心绝对构型通过X射线晶体学结构测定确证。商业化工艺生产得到的原料药为单一晶型X,为BCS 4类。原料药粒度大小和晶型并不是关键质量属性。
申请人采用了成熟的原料药合成工艺,该工艺根据ICH指导原则充分控制了残留溶剂、元素杂质及致突变杂质,起始物料设定合理。原料药质量控制标准包含了性状、红外光谱和高效液相色谱法鉴别、含量测定、手性纯度、水分、有机杂质及微生物限度等关键质量属性。用于控制原料药质量的分析方法充分且经过适当验证。稳定性数据可以用于支持拟定条件下的储存时间。
Crinecerfont原料药为低溶解低渗透性化合物,符合BCS 4类特征。不同于多数原料药的质量控制,Crinecerfont原料药的质量标准中控制了微生物限度。
制剂部分评估
从药理/毒理学以及CMC角度来看,制剂中新型辅料(即丙二醇二辛酸酯)的含量水平被认为是适宜的。制剂中有固定比例的已知辅料,3期和商业化制剂对比时,其比例没有变化。
原料药粒径预计不影响产品质量。在产品开发过程中,已确定关键质量属性,并通过包含性状、鉴别、含量测定、降解产物、溶出度、剂量单位均匀性和微生物属性等项目检测产品,进行了充分控制。用于产品质量控制的分析方法均充分且经过适当验证。根据提供的稳定性数据,申请方拟定的15个月有效期(储存条件15°C - 25°C/59°F至77°F)可被接受。
药品装在一个120cc的高密度聚乙烯瓶中,瓶口上有一个38毫米的儿童防护帽和铝箔密封。
Crinecerfont软胶囊处方中使用了一种新型辅料丙二醇二辛酸酯,评估了其在制剂中的含量水平,安全性可控。企业申请了15个月的有效期。储存条件为15°C - 25°C,不同于一般固体制剂的USP要求:20°C - 25°C 常见储存条件。
生物药剂学:
Crinecerfont软胶囊,规格为25 mg、50 mg和100 mg,是一种选择性促肾上腺皮质激素释放因子1(CRF1)受体拮抗剂。该药物用于4岁及以上患者的推荐剂量:应随餐服用,每日两次(早晚各一次)。
FDA审评员认为Crinecerfont是一种溶解性差的化合物。在质量控制上,申请人拟定的两阶段溶出方法是充分的,总体生物药剂学风险较低。FDA认为三种规格的胶囊配方在组成成分上呈现比例一致性。基于提交的关于处方间桥接、新增规格、生产场地及批量的整体性信息与数据,根据21 CFR 320.22(d)(2)法规要求,针对"拟上市"处方中25 mg和50 mg规格豁免体内生物等效性研究证明的要求具有可接受性,无需开展额外的体内研究。
(i)体外溶出方法的开发、体外溶解数据和体外溶解接受标准;
(ii)临床和商业/上市药物产品之间的桥接数据、制造地点和批次大小;
(iii)拟定低规格药物的生物豁免请求。
体外溶出方法及验收标准:
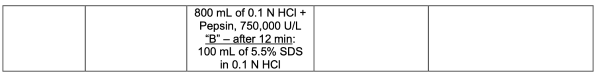
本品含低溶解性药物活性成分,采用中链甘油三酯、丙二醇、月桂酰聚氧乙烯甘油酯及维生素E聚乙二醇琥珀酸酯(维生素E TPGS)作为溶剂配制成溶液。申请人提出了一种两阶段溶出方法,在第二阶段溶出方法中,介质中使用胃蛋白酶的方法被认为是可接受的。所提出的溶出方法CFVs/CPPs/ CMAs变化(针对某辅料浓度的改变)具有有限的区分能力,经评估被认为可以接受。
基于溶出两阶段的接受限度,拟定的标准是可以接受。经申请人和FDA达成的一致同意,用于Crinecerfont软胶囊(25 mg、50 mg和100 mg)在批放行及稳定性研究期间质量控制的最终溶出度方法和验收标准如下表1所示。

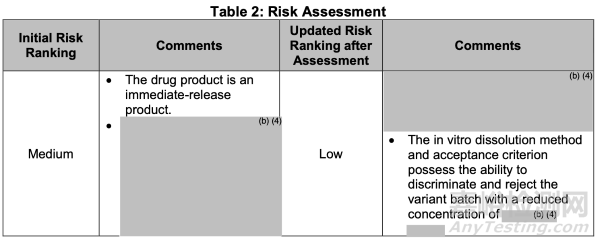
从生物药剂学角度,基于对关键制剂变量(CFVs)/关键生物利用度属性(CBAs)的识别,初始风险等级被评估为"中"。该评估基于分装单元操作已建立并维持所有适用的GMP规范的假设。通过风险缓解及控制策略的实施,最终风险等级已更新为"低"(见表2)。
企业拟定的商业化溶出方法为两阶段溶出,第1阶段使用0.6%SDS表面活性剂;第2阶段前12分钟使用了胃蛋白酶,12分钟后增加了100ml 5.5%的SDS,Q点设定为45分钟,使用了高浓度表活和高浓度胃蛋白酶,可预见原料药溶解度是难溶化合物。
低规格(25 mg和50 mg)的桥接:
针对100 mg规格的拟商业化(TBM)制剂进行了一项1期研究。根据21 CFR 320.22(d)(2)规定,为支持25 mg和50 mg规格的TBM制剂与100 mg规格TBM制剂的桥接,申请人提交了数据证明。
1.单次口服Crincefont后,其暴露参数(Cmax和AUC)在胶囊制剂的临床相关剂量范围(25至200 mg)内呈剂量比例关系。
2.TBM胶囊100 mg、50 mg和25 mg规格的填充制剂生产工艺及定性与定量组成均保持比例一致性。
3.用于体外对比的参比制剂(100 mg TBM胶囊)已在体内临床研究中证明与III期胶囊具有生物等效性。
4.TBM 100 mg胶囊(1×100 mg)与注册批25 mg胶囊(4×25 mg和1×25 mg[标示量%])、50 mg胶囊(2×50 mg和1×50 mg[标示量%])的体外溶出曲线比较数据(f2值)。
企业在临床1期使用了100mg规格制剂,对于其他拟上市规格25mg和50mg,对比了与100mg规格之间相似性,包括:25mg和50mg规格暴露剂量与100mg规格呈现剂量比例关系;对比100mg处方,25mg和50mg规格是等比例处方。而且25mg,50mg和100mg三个规格在等浓度的溶出试验中呈曲线呈相似性。因100mg已证明与确证性临床胶囊等效,因此上述三个规格在体内的等效性可桥接。
生产场地的桥接:
拟商业(TBM)三种规格批次的制造均使用单一生产场地。TBM产品的100毫克规格已用于临床1期研究。因此无需进行生产场地桥接。
批次规模的桥接:
拟商业化代表批/临床批次三种规格的生产处方均为软胶囊剂型。由于各代表批/临床批次规模差异在10倍范围内,基于I期研究数据,代表性/临床批次进行桥接被认为可接受。拟上市商业化批次的三种规格规模,根据SUPAC-IR指南,由于代表性/临床批次与拟上市商业批次规模差异也在10倍范围内,建议通过对比代表性/临床批次与拟上市商业批次的体外溶出曲线,即可建立商业批次与临床批次的桥接关系。
拟上市三规格的批量规模与确证性临床批次的批量规格在10倍以内,因此不需要进行体内数据桥接,仅需进行制剂体外的溶出曲线对比,参考上述的溶出数据,可认为拟上市三个规格的批量规模与确证性临床批次的规格是可桥接的。
生物等效性豁免申请:
基于桥接不同制剂、新增规格、生产场地及批量大小所提交信息的整体性,支持对拟商业化制剂25 mg和50 mg规格免于体内生物等效性研究的依据,符合21 CFR 320.22(d)(2)要求,被认为是可接受的。无需进行额外的体内研究。25 mg与50 mg规格的疗效研究将由临床/临床药理学审评员进行评估。
因此,拟商业化制剂25 mg和50 mg规格的生物等效性豁免申请得到了FDA的同意,基于制剂处方、场地、批量和暴露量等核心数据的可桥接性。

来源:文亮频道